
eBook - ePub
Facioscapulohumeral Muscular Dystrophy (FSHD)
Clinical Medicine and Molecular Cell Biology
- 250 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Facioscapulohumeral Muscular Dystrophy (FSHD)
Clinical Medicine and Molecular Cell Biology
About this book
Facioscapulohumeral muscular dystrophy (FSHD) is a genetic disorder involving slowly progressive muscle degeneration in which the muscles of the face, shoulder blades and upper arms are among the most severely affected. It is the third most common inherited muscular dystrophy, affecting 1 in 20,000. The search for the molecular basis of the disease is of interest to all genetic researchers, involving a deletion outside a coding region resulting in over-expression of adjacent genes. This volume summarizes the current understanding of the disorder, including clinical, molecular and therapeutic aspects.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Topic
MedicineSubtopic
Cardiology1. Introduction and overview of FSHD
Meena Upadhyaya, and David N.Cooper
FSHD Facioscapulohumeral Muscular Dystrophy: Clinical Medicine and Molecular Cell Biology, edited by Meena Upadhyaya and David N.Cooper. © 2004 Garland/BIOS Scientific Publishers Limited, Abingdon.
1.1 Introduction
FSHD is a unique genetic disorder. For a long time, the molecular basis of this condition was enigmatic. Indeed, some 13 years elasped between the mapping of the chromosomal location of the gene and deducing the nature of the unique molecular mechanism underlying the disease. The last decade has thus been an extremely exciting one for FSHD research (Figure 1.1). During this time it is clear that there has been a very significant increase in our knowledge of the molecular genetics of FSHD.
Facioscapulohumeral muscular dystrophy (FSHD) is the third most common inherited neuromuscular condition, after Duchenne and myotonic muscular dystrophies (Upadhyaya and Cooper, 2002). FSHD is an autosomal dominant inherited disorder characterized by progressive muscle weakness and involving atrophy of the muscles of the face, upper arm and shoulder girdle (see Chapters 2 and 3). General muscle weakness and atrophy may eventually involve musculature of the pelvic girdle and the foot extensor. Early onset of the disease is usually associated with the development of the most severe forms of the disorder (Lunt and Harper, 1991; Padberg, 1998). Disease onset is unusual before the age of 10, however, being observed in fewer than 5% of patients, in whom it is associated with significant facial weakness. The majority of patients only develop symptoms later during the second decade of life. Both retinal vasculopathy and high-tone deafness may be seen as part of FSHD (see Chapter 13). However, clinically significant deafness is rare in adult FSHD. In some families, the affected individuals share many of the clinical features of FSHD but show no evidence of facial muscle involvement, even in the most severely affected patients. These ‘scapulohumeral muscular dystrophy’ families display an autosomal dominant mode of inheritance, and it has been suggested that they may form part of the FSHD disease spectrum (Jardine et al., 1994; Tawil et al., 1995; Felice et al., 2000). In some FSHD families, the disease is associated with mental retardation and epilepsy (see Chapter 14).
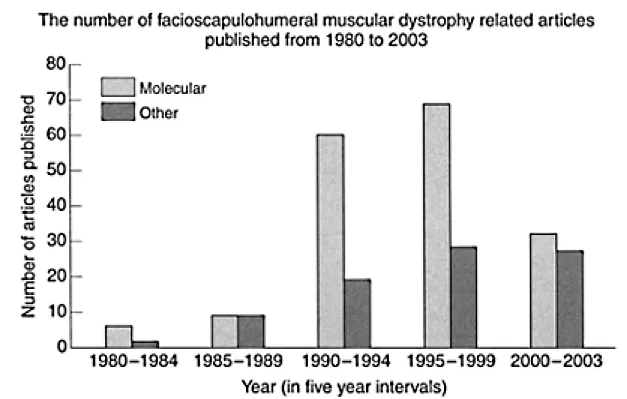
Figure 1.1 The total annual number of publications relating to FSHD retrieved from PubMed (NCBI), from 1980.
1.2 Gene mapping studies
The FSHD locus was genetically mapped to chromosome 4q35 (Upadhyaya et al., 1990; Wijmenga et al., 1990) where it is closely linked to the complex locus D4F104S1 (Figure 1.2) identified by Southern analysis using probe p13E-11 (Wijmenga et al., 1992). Details of the mapping studies are given in Chapter 4. There is some evidence for genetic heterogeneity in FSHD. Although the vast majority of large FSHD families can be demonstrably linked to 4q35, a small number of classical FSHD families have been excluded from this location (Gilbert et al., 1993). A genomic position for this second disease locus, tentatively designated FSHD1B, has still to be identified (see Chapter 19).
1.2.1 DNA rearrangements associated with D4F104S1 and D4Z4 repeats
Probe p13E-11 (D4F104S1) identifies an EcoRI restriction fragment, with homology to both 4q35 and 10q26. In FSHD patients, one fragment derived from one of the two homologous chromosomes 4, is usually smaller than 35 kb. Studies in suitably informative FSHD families have demonstrated the 4q35 component of the D4F104S1 locus to be the closest marker to the disease gene. Sequence analysis of the entire D4F104S1 locus has shown that p13E-11 identifies a single copy genomic sequence that is located immediately proximal to an array of tandem repeat units. It is variation in the exact number of individual repeats, in each of the 4q35- and 10q26-derived repeat arrays, that constitutes the polymorphic element of the D4F104S1 variable number tandem repeat (VNTR) and of the two specific EcoRI restriction sites which define this VNTR; one site is located proximal to the sequence homologous to the p13E-11 probe, whereas the second is situated immediately distal to the tandem repeat array (see Figure 1.2).
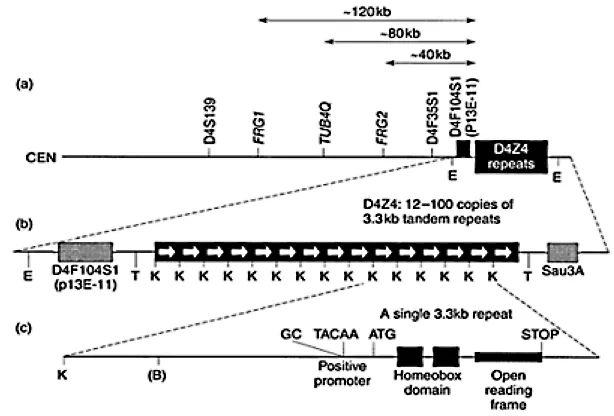
Figure 1.2 Restriction map of the FSHD candidate region at 4q35: Relative positions of FRG1, TUBB4Q, FRG2, D4F104S1 (p13E11) and D4Z4. The EcoRI fragment detected by probe p13E11 predominantly comprises an array of 3.3 kb tandem repeats that have a copy number of 12–100 in normal controls and usually <10 in FSHD patients. Each 3.3 kb repeat comprises two homeodomains (shaded boxes) encompassing an ORF with an in-frame start codon (ATG) and a stop codon. It also encodes the DUX4 gene. The position of the GC and TACAA boxes in the promoter-like sequence of DUX4 gene is indicated. B: BlnI restriction site, E: EcoRI restriction site; K: KpnI restriction site; T: Tru91 restriction site.
Each (virtually identical?) repeat unit is 3.3 kb in size and has been designated D4Z4, regardless of whether the repeat is located within the 4q35- or the 10q26-derived repeat array. The sizes of the variable fragments seen on Southern blots, containing EcoRI-digested high-molecular-weight genomic DNA derived from normal individuals and hybridized with probe p13E-11, range from ~35–38 kb to more than 300 kb whereas in FSHD patients, they range from 7 to 35–38 kb (see Chapter 4). Since it is difficult to resolve fragment sizes over 50 kb with conventional Southern blots, pulsed-field gel electrophoresis (PFGE) has been employed to increase resolution. Since the majority of people carry two copies of each homologous chromosome, there is, in an informative situation, the potential to observe four differently sized EcoRI fragments when the DNA from one individual is analysed. Each fragment represents a different D4F104S1 allele, with two alleles being derived from the 4q35 locus and two alleles from the 10q26 locus (both loci produce EcoRI fragments with the same size range). Following such analyses in a number of FSHD families, it was soon realized that the size of at least one of the four D4F104S1 alleles seen in the affected members of these families, differed significantly from the normal size range: patients possessed EcoRI fragments in the range from ~35 kb down to ~10 kb.
This potential disease association was confirmed and strengthened by demonstrating the de novo occurrence of a smaller EcoRI fragment in the majority of sporadic FSHD patients studied, that was not present in either of the clinically unaffected parents. A small proportion of FSHD patients, of both sporadic and familial origin, do not exhibit this EcoRI fragment, either because (i) such cases arise via a different mutational mechanism, (ii) they represent families unlinked to the FSHD1A locus at 4q35, or (iii) they are phenocopies of other conditions. What pathognomonic process could turn this apparently normal variation into a diseasecausing mutation? One clue is perhaps to be found in the observation that the severity of disease expression in FSHD patients correlates with the size of their disease-associated D4F104S1 alleles. Thus, patients with the smallest EcoRI fragments manifest the severe form of the disorder and present with an earlier age of onset (Lunt et al., 1995).
If the possession of one short D4F104S1 allele is sufficient to confer the disease phenotype (FSHD is a dominant disorder, and hence, only requires a change in one of the two copies of the FSHD gene), then one might infer that the complete loss of one of the D4F104S1 alleles would yield a still more severe phenotype. A number of individuals have been identified who carry small cytogenetically detectable deletions involving specifically the 4q region and, as a result, are monosomic for one copy of their 4q35- derived D4F104S1 loci. Surprisingly, none of these individuals showed any evidence of an FSHD-like disorder; indeed, most of them appeared to be phenotypically normal. These findings indicated that FSHD most probably results from a dominant negative (‘gain-of-function’) mutational mechanism rather than the loss or partial loss of a gene product (haploinsufficiency) from within the 4q-deleted region. These observations served to focus the search for the FSHD gene back onto the D4F104S1 locus itself, and in particular to the D4Z4 repeats therein.
1.2.2 D4Z4 repeats
The D4Z4 repeat units are members of a large family of 3.3 kb tandem repeat loci that are located on the short arm of the acrocentic chromosomes, the pericentromeric regions (especially on chromosome 1), and the telomeric regions of the long arms of chromosomes 4 and 10 (Hewitt et al., 1994; Lyle et al., 1995). Chapter 6 describes the structural organization and evolution of the D4Z4 repeats. The size variation observed at the D4F104S1 locus is caused by the loss of individual discrete D4Z4 repeat units and does not involve internal deletions within the body of the repeat itself (van Deutekom et al., 1993). Thus, the normal size range of the D4F104S1 alleles, from about 38 kb to more than 300 kb, represents D4Z4 repeat arrays containing from 11–100 repeats. By contrast, the FSHD-associated D4F104S1 allele size range, from 10 kb to ~38 kb, represents arrays containing between one and 11 D4Z4 repeat units. Large-scale physical mapping and sequencing studies of the entire D4F104S1 genomic region have demonstrated: (i) that the 4q35 D4Z4 arrays are located immediately adjacent to the 4q telomere; and (ii) that a copy of a single D4Z4 repeat-like sequence is located about 30 kb proximal to the p13E-1 1 homologous sequence in the opposite orientation to the D4Z4 repeats in the distally located tandem arrays.
1.2.3 Sequence homology and genetic recombination between 4q35 and 10q26
Various studies have shown that a duplicate (virtually identical) copy of the D4F104S1 locus is located at 10q26, within the heterochromatic subtelomeric region of chromosome 10 (Deidda et al., 1995). Detailed sequence analysis of D4Z4 repeats from both the 4q35 and 10q26 D4F104S1 loci confirmed their high level of sequence homology (98–100%) and, perhaps more importantly, identified a unique BlnI restriction site that is present within each copy of the 10q26-derived repeat units, but which is absent from the 4q35- derived D4Z4 repeats (Deidda et al., 1996).
The high degree of sequence homology between the 4q35 and 10q26 D4F104S1 regions may be responsible for the observed ‘interchromosomal exchanges’ between these two loci (see Chapter 7). During these subtelomeric exchanges, complete repeat arrays (either of 4q35-derived BlnI-resistant D4Z4 repeats, or of 10q26-derived BlnI-sensitive repeats) are ‘transferred’ from one chromosomal location to the other. Owing to the sizes of the genomic fragments involved, these interchromosomal exchange events are best visualized with PFGE; such studies have shown that entire repeat arrays are ‘translocated’ in the majority of studied cases (van Deutekom et al., 1996a). It should however be stated that there is no direct evidence for the actual physical translocation of genetic material and interchromosomal gene conversion would perhaps appear to be a more plausible mechanism.
Perhaps surprisingly, these dynamic subtelomeric interchanges do not appear to be involved in any way with expression of the FSHD phenotype. Such 4q35 10q26 exchanges are evident in about 20% of normal individuals. It should be emphasized, though, that FSHD only occurs when the D4Z4 repeats that are deleted (regardless of whether they are originally chromosome 4q35- or 10q26-derived) are chromosome 4 located (van Deutekom et al., 1996a). Recently, it has been demonstrated that in FSHD patients, homogeneous chromosome 10-specific repeats are rarely encountered. Rather, these repeats are invariably combined with chromosome 4-specific repeats (van Overveld et al., 2000). The molecular mechanism(s) underlying these subtelomeric exchanges has still to be elucidated, as has their potential influence (if any) on FSHD disease expression.
1.3 Somatic mosaicism
Somatic mutations that occur sufficiently early on in embryonic life involve both somatic cells and the germline. These individuals (gonosomal mosaics) may also be at risk of having affected children. Often a new mutation first appears in mosaic form, usually in a clinically normal person, who then has a constitutionally affected child. Somatic mosaicism in FSHD has been reported (Upadhyaya et al., 1995; Kohler et al., 1996). van der Maarel et al. (2000) detected somatic mosaicism in 40% of de novo FSHD families (14% in an unaffected parent and 26% of the de novo FSHD patients themselves). Interestingly, an excess of mosaic-affected males was found in this dataset. Chapter 12 describes mosaicism in FSHD.
1.4 The genotype/phenotype relationship in FSHD
An association between the size of the deleted EcoRI fragment and the age at disease onset (smaller EcoRI fragments are always associated with the most severe form of the disease) has been observed in patients from a number of different ethnic groups (Lunt et al., 1995; Zatz et al., 1995; Tawil et al., 1996; Hsu et al., 1997). The D4F104S1 EcoRI fragment size range noted in severe childhood cases is 10–18 kb, in typical teenage-onset cases between 18–34 kb, and in the oldest late-onset patients larger than 30 kb (Lunt et al., 1995; see Chapter 11). Two recent Japanese studies have confirmed these observations; a number of very-early-onset patients, with a severe form of FSHD often accompanied by epilepsy and mental retardation, manifested short EcoRI/BlnI fragments, the smallest being 10 kb in length (Funakoshi et al., 1998; Muira et al., 1998). Chapter 14 gives an account of unusual clinical features in FSHD. Genotype/phenotype correlations do not however always appear to be sustained in the borderline region of 8–14 D4Z4 repeats (Butz et al., 2003).
It is important to stress that it is not yet possible to predict accurately the likely severity of disease expression in any one individual. This is due to the high degree of inter- and intrafamilial variability of disease expression observed in this disorder, regardless of the fact that all affected members of a family exhibit the same size of D...
Table of contents
- Cover Page
- Title Page
- Copyright Page
- Abbreviations
- Contributors
- Acknowledgements
- Dedication
- Foreword
- 1. Introduction and overview of FSHD
- 2. Facioscapulohumeral muscular dystrophy: historical background and literature review
- 3. Facioscapulohumeral muscular dystrophy: a clinician’s experience
- 4. Mapping of the FSHD gene and the discovery of the pathognomonic deletion
- 5. Identification and characterization of candidate genes in the FSHD region
- 6. Evolution and structural organization of the homeobox-containing repeat D4Z4
- 7. Subtelomeric exchange between 4q and 10q sequences
- 8. Genomic analysis of the subtelomeric regions of human chromosomes 10q and 4q: relevance to FSHD
- 9. The DUX gene family and FSHD
- 10. Facioscapulohumeral muscular dystrophy (FSHD): a disorder of muscle gene derepression
- 11. Genotype-phenotype relationships in FSHD
- 12. Mosaicism and FSHD
- 13. Retinal vascular abnormalities in FSHD: a therapeutic message; clues to pathogenesis?
- 14. Unusual clinical features associated with FSHD
- 15. Molecular diagnosis of FSHD
- 16. FSHD myoblasts: in vitro studies
- 17. Exploring hypotheses about the molecular aetiology of FSHD: loss of heterochromatin spreading and other long-range interaction models
- 18. Histological, immunocytochemical, molecular and ultrastructural characteristics of FSHD muscle
- 19. Linkage analysis in non-chromosome 4-linked FSHD
- 20. Facioscapulohumeral muscular dystrophy: gender differences and genetic counselling in a complex disorder
- 21. Genetic counselling for facioscapulohumeral muscular dystrophy (FSHD)
- 22. Sarcolemmal reorganization in FSHD
- 23. Expression profiling in FSHD
- 24. Therapeutic trials and medical management in FSHD
- Appendix I The FSH Society
- Appendix II The Muscular Dystrophy Campaign: pioneering research, providing care
- Appendix III The Association Française contre les Myopathies
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Facioscapulohumeral Muscular Dystrophy (FSHD) by David Cooper,Meena Upadhhyaya in PDF and/or ePUB format, as well as other popular books in Medicine & Cardiology. We have over 1.5 million books available in our catalogue for you to explore.