
- 215 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
About this book
Noonan Syndrome: Characteristics and Interventions provides an in-depth analysis on this disorder that pediatric endocrinologists and primary care clinicians can use to make sure they provide affected patients with an updated model of care and appropriate treatment. The book examines recent advances in understanding and treating short stature in Noonan Syndrome, along with the latest progress in growth hormone-dependent signaling pathways involved in short stature, one of the most frequent clinical manifestations. Chapters also address how patients with Noonan Syndrome undergo more than average surgical procedures and have a great bleeding risk.
This must have reference for pediatric endocrinologists and practicing physicians will give them all the information they need on the topic.
- Provides an accessible, up-to-date overview of the characteristics, state-of-the-art diagnostic procedures, and management of Noonan syndrome
- Offers an important resource for physicians who see and treat individual symptoms, rather than a disease complex, covering the important characteristics in the presence of heart anomalies and perioperative considerations
- Reviews multidisciplinary and post-treatment management of the disease
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Topic
MedicineSubtopic
Endocrinology & MetabolismChapter 1
Noonan Syndrome: Phenotypic Variations and Molecular Genetics
Neda Zadeh Genetics Center, Orange, CA, United States
CHOC Children's Hospital, Orange, CA, United States
CHOC Children's Hospital, Orange, CA, United States
Abstract
Noonan syndrome is a genetic multisystemic disorder with a prevalence of 1 in 1000–2500 newborns. This condition is characterized by dysmorphic features, developmental delay, short stature, congenital heart disease, lymphatic malformations, genitourinary anomalies, and bleeding difficulties. Mutations that cause Noonan syndrome alter genes encoding proteins with a role in the RAS/MAPK pathway. Medical management guidelines have been developed for this condition and molecular genetic testing is available diagnostically and may provide further aid in long medical management and prognosis (Roberts et al. [5]).
Keywords
Noonan syndrome; RASopathy; Ras-MAPK; PTPN11
Abbreviations
CFC cardiofaciocutaneous syndrome
NS Noonan syndrome
NT nuchal translucency
RAS/MAPK pathway Ras/mitogen-activated protein kinase pathway
Introduction
Noonan syndrome (NS) is a multisystemic genetic condition that occurs in 1:1000–2500 live births. An incorrect term utilized to describe this condition is the “male Turner syndrome” which incorrectly implied that this condition would not be observed in females. As Noonan syndrome is an autosomal dominant disorder, it is observed in both males and females [1]. Characteristic features include short stature, congenital cardiac defects, dysmorphic features, and variable developmental delay and learning difficulties. Other features include sternal abnormalities, genitourinary abnormalities, lymphatic dysplasia, and coagulation defects. Hypertrophic cardiomyopathy can also be observed in approximately 20%–30% of patients. In addition, individuals with Noonan syndrome have an increased risk of developing cancer. Noonan syndrome is diagnosed clinically, with the average age at diagnosis of 9 years [2]. Molecular testing is becoming much more utilized to provide diagnostic confirmation as well as to attempt to provide genotype-phenotype correlation. Mutations that cause Noonan syndrome alter genes encoding proteins within the RAS/MAPK pathway, which plays a role in a variety of functions including proliferation, migration, cell fate determination, and senescence [3]. This pathway also participates in early and late developmental process including organogenesis, morphology determination, and growth [4]. Heterozygous pathogenic mutations in nine genes account for approximately 75%–80% of all Noonan syndrome cases. Within the past several years, whole exome sequencing has allowed identification of new variants in rare genes [3]. Therefore molecular diagnosis is helpful in diagnostic confirmation and also potentially provides genotype-phenotype correlations and accurate risk assessment [1,5].
RASopathies
The RAS/MAPK pathway is an important signal transduction pathway through which extracellular ligands stimulate cell proliferation, differentiation, survival, and metabolism [5] (Fig. 1). Ligand binding to cell surface receptors causes site-specific phosphorylation within certain cytoplasmic regions. This leads to recruitment of adaptor proteins which form a complex with guanine nucleotide exchange factors that convert inactive RAS-GDP to its active RAS-GTP form. This activated form of RAS protein then activates the RAF-MEK-ERK cascade through a series of phosphorylation events. The end product of activated ERK then enters the nucleus to alter gene transcription [5]. Due to its role in signal transduction, signal through the RAS/MAPK pathway is normally tightly controlled, with enhanced flow through the pathway contributing to oncogenesis [4].

The RASopathies are a clinically defined group of medical genetic syndromes caused by mutations in genes that encode components or regulators of the RAS/MAPK pathway as described before [6]. Genetic conditions included in the group of RASopathies include Noonan syndrome, Costello syndrome, cardiofaciocutaneous (CFC) syndrome, Legius syndrome, Noonan syndrome with multiple lentigines (formerly LEOPARD syndrome), and capillary malformation-ateriovenous malformation syndrome (CM-AVM) [6].
All the known genes associated with Noonan syndrome encode proteins integral to the above described pathway [5,6]. As the other RASopathy conditions are associated with genes that are integral to the function of the RAS/MAPK pathway, many clinical features may be similar or overlapping, often making clinical diagnoses difficult and underlying the diagnostic aid of molecular testing.
Clinical Features of Noonan syndrome
Noonan syndrome should be suspected in individuals with the following key features (modified from Allanson et al. [1]):
- • Characteristic facial features. Facial features of patients with Noonan syndrome show considerable differences depending on the age of the patient, being more striking during infancy and adolescence, and more subtle in adulthood [2] (Fig. 2).
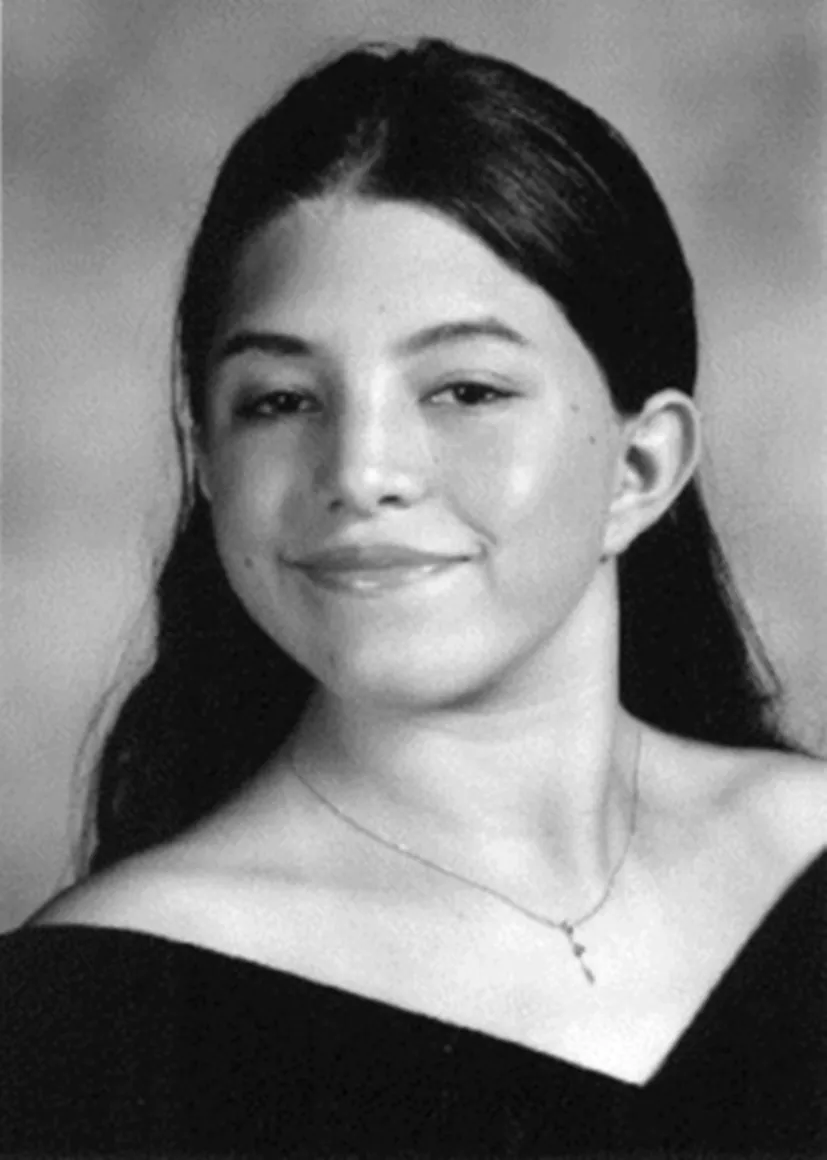
Fig. 2 Eighteen-year-old young lady with Noonan syndrome. (Reproduced from Noonan JA. Noonan syndrome and related disorders. Prog Pediatr Cardiol 2005;20(2):177–85. https://doi.org//10.1016/j.ppedcard.2005.04.008, Elsevier.)
During infancy, the head can appear relatively large with a tall forehead, hypertelorism with downslanting palpebral fissures, low-set posteriorly rotated ears with thickened helix, deeply grooved philtrum with high wide peaks to the vermillion border of the upper lip [1,2,5]. Eyes are often prominent with full or ptotic eyelids. There is usually a depressed nasal root, wide base, and bulbous tip. The hair can by wispy during the toddler years [1,2].
In childhood there is often myopathic facies with decreased facial expression. By adolescence the facial shape resembles an inverted triangle with a wide forehead and tapered chin. Eyes often become less prominent, and the neck lengthens, which may accentuate nuchal webbing. The hair is often curly or wooly [2].
In adults there are prominent nasolabial folds and skin can often have a transparent and prematurely wrinkled appearance [1,2].
Ocular anomalies can include strabismus, refractive errors, amblyopia, and nystagmus in up to 95% of patients. There are a few case reports of keratoconus and Axenfeld anomaly. Iris color can be lighter than what is typically expected for ethnic background [1,2]. - • Short stature. Birth weight and length are generally normal with signs of postnatal growth failure noted by 12 months of age, which tends to track along the 3rd percentile from early childhood until puberty. The prevalence of short stature in Noonan syndrome is most notable during pubertal ages, often with delayed bone age. Often, catch-up growth can occur during later teen years, with prolonged growth into the 20s that is possible. Growth hormone deficiency, neurosecretory dysfunction, and growth hormone resistance have been described. Growth hormone treatment data can be difficult to compare due to differing protocols and outcome criteria [5]. However, short stature due to Noonan syndrome is an FDA-approved indication for growth hormone treatment. The final adult height can approach the lower limit of normal with 161–167 cm in males and 150–155 cm in females [1,2]. Specific growth curves exist and can be accessed under Noonan syndrome clinical management guidelines on the RASopathies network web page (https://rasopathiesnet.org).
- • Congenital cardiac defect. Noonan syndrome is the second most common syndromic cause of congenital heart disease [7]. The most common abnormalities include pulmonary valve stenosis often with dysplasia (20%–60%), hypertrophic cardiomyopathy (20%–30%), and secundum atrial septal defect (6%–10%). Ventricular septal...
Table of contents
- Cover image
- Title page
- Table of Contents
- Copyright
- Contributors
- Author Biography
- Foreword
- Chapter 1: Noonan Syndrome: Phenotypic Variations and Molecular Genetics
- Chapter 2: Growth Failure and Experience With Growth Hormone Therapy in Noonan Syndrome
- Chapter 3: Cardiac Manifestations in Noonan Syndrome: Effects of Growth Hormone Therapy
- Chapter 4: Endocrinopathies Associated With Noonan Syndrome
- Chapter 5: Genitourinary Manifestation of Noonan Syndrome
- Chapter 6: Gastrointestinal Manifestations of Noonan Syndrome
- Chapter 7: Developmental and Neurological Features of Noonan Syndrome
- Chapter 8: Hematology/Oncology in Noonan Syndrome
- Chapter 9: Analgesia, Anesthesia, and Perioperative Considerations in Noonan Syndrome
- Chapter 10: Oral and Dental Manifestations in Noonan Syndrome
- Management of Noonan Syndrome: A Clinical Guideline: Noonan Syndrome Guideline Development Group
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Noonan Syndrome by Amrit P.S. Bhangoo in PDF and/or ePUB format, as well as other popular books in Medicine & Endocrinology & Metabolism. We have over 1.5 million books available in our catalogue for you to explore.