![]()
Part I
Introduction
![]()
Introduction
Downstream biomanufacturing processes increase product concentration and purity, while decreasing process volume. Therefore, decreasing process volume without loss of product is essential to increase product purity, while at the same time eliminating product contaminants. The biochemistry of different products (peptides, proteins, hormones, low-molecular-weight metabolic intermediates, complex antigens etc.), all of which are liable to degradation, dictates that different separation methods be used to isolate and purify these products from contaminating biomolecules produced by the upstream process. Optimal downstream product yield is the yield of recovered product in the appropriate final biologically active form and purity. Purified but inactive product is a contaminant, reduces overall process yield, and may have serious consequences on clinical safety and efficacy. That is why downstream process design has the greatest impact on the overall biomanufacturing cost.
As product purity increases, more product can be lost to inactivation, nonspecific binding to equipment surfaces, binding to membranes, and chromatography media or by precipitation, thus decreasing the recovery of product. Because of these potential losses, each additional separation step may reduce overall yield. Therefore, downstream separation scientists and engineers are continually seeking to eliminate or combine unit operations to minimize the number of process steps in order to maximize product recovery at a specified concentration and purity.
Section II of Downstream Industrial Biotechnology includes detailed methods used for the initial steps of cell separation, cell disruption (for intracellular products), filter aids and adsorbents for rapid protein capture and initial volume reduction. Each of these steps is critically affected by upstream process design (volume, product concentration, and contaminants derived from the growth media or host cells), which impacts every subsequent step of downstream product recovery and purification. In particular, cell separation and cell disruption methods can have a dramatic effect on contributing (or minimizing) contaminants such as nucleic acids, host cell proteins, cell membrane fragments or pyrogenic lipopolysaccharides that need to be removed from the final product in subsequent separation steps.
Although each upstream process decision impacts downstream product recovery and purification, not all contaminants come from upstream operations. In some cases contaminants can also be generated by downstream operations, as inactivated product (due to heating, proteolysis, photoinactivation or precipitation), bioburden or microbial contamination introduced during downstream operations (from the environment, water, operations staff etc.) or contaminants derived from materials in direct contact with the product (extractable, leachable contaminants).
The downstream steps described in Section III are optimized by absorbent surface area, selectivity, binding capacity, and degree of volume reduction to purify product in the concentration range needed for each subsequent step to meet overall criteria of scale, stability, purity, and potency. Therefore, close integration of the characteristics of the upstream biological system that produces the product with the engineering and optimal performance of the downstream product separation, concentration. and purification operations are essential. This means that separation engineers, bioseparation and bioanalytical scientists, and manufacturing operations staff with broad expertise in working with labile biological molecules all need to work and communicate effectively as a team to design a downstream process that can be scaled from the laboratory bench and transferred to the manufacturing scale. It also means that downstream process scientists must continually provide feedback information to upstream process engineers and scientists to minimize the impact of upstream changes (cell line changes, media composition changes, the addition of antifoam, degradation of product during in-process storage or holds) on downstream separation operations. Therefore, the companion volumes of Upstream Industrial Biotechnology should also be consulted when designing a downstream process.
Each downstream step requires process development and optimization (for purity, overall yield) because of the complexity of the structure of the biological molecules being purified and the complexity of contaminants. Section III also includes approaches for scale down of purification operations. Each downstream step is expensive to optimize at the pilot or manufacturing scale. This expense is not only due to the scale of the equipment and expense of the separation media, but also because of the large quantity of valuable product needed to carry out optimization studies at scale.
Downstream operations require specialized equipment designed for separation of proteins, peptides, virus, particulate antigens or low-molecular-weight biomolecules while minimizing product degradation. Sections IV and V focus on large scale equipment design and fluid transfer systems, and describe in detail many types of industrial bioseparation equipment. Of particular concern for products derived from mammalian cell lines are effective methods for virus inactivation and viral filtration that can be validated with model virus challenge. These methods are described in section V.
Not only do the upstream and downstream processes need to be designed to meet cGMPs and be capable of being licensed, but the facility used to carry out the process also must be designed so that it can be licensed. Section VI and VII of Downstream address facility design, facility validation, clean-in-place (CIP) and sterilization-in-place (SIP) methods. A major advance in facility design for downstream processes is the growing impact of single use (SU) disposable downstream materials and this is described in Section VI.
The overall goal of all downstream operations is not only to purify bulk product for formulation, but to achieve regulatory compliance and licensure so that final formulated and filled product can be released to consumers, physicians or patients. Section VII describes how Process Analytical Technology (PAT), bioburden testing and Quality by Design (QbD) impact downstream process design and contribute to regulatory compliance both for the USFDA and European regulatory agencies.
![]()
Chapter 1
Bioprocess Design, Computer-Aided
Victor Papavasileiou
Intelligen Europe, Leiden, The Netherlands
Charles Siletti
Intelligen, Inc., Mt. Laurel, New Jersey
Alexandros Koulouris
Intelligen Europe, Thermi, Greece
Demetri Petrides
Intelligen, Inc., Scotch Plains, New Jersey
1.1 Introduction
Bioprocess design is the conceptual work done prior to commercialization of a biological product. Given information on the potential market demand for a new product, bioprocess design endeavors to answer the following questions: What are the required amounts of raw materials and utilities for manufacturing a certain amount of product per year? What is the required size of process equipment and supporting utilities? Can the product be manufactured in an existing facility or is a new plant required? What is the total capital investment for a new facility? What is the manufacturing cost? How long does a single batch take? What is the minimum time between consecutive batches? During the course of a batch, what is the demand for various resources (e.g. raw materials, labor, and utilities)? Which process steps or resources are the likely production bottlenecks? What process and equipment changes can increase throughput? What is the environmental impact of the process? Which design is the “best” among several plausible alternatives?
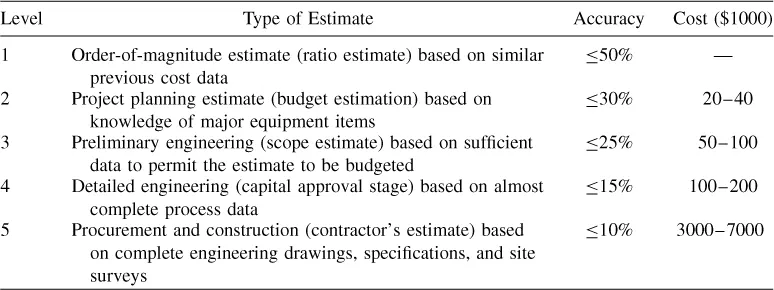
Bioprocess design and project economic evaluation require the integration of knowledge from many different scientific and engineering disciplines. Design and evaluation are also carried out at various levels of detail. Table 1.1 presents a common classification of design and cost estimates and typical engineering costs for a $50 million capital investment project (1).
Table 1.1 Types of Design Estimates
Order-of-magnitude estimates are usually practiced by experienced engineers who have worked on similar projects in the past. They take minutes or hours to complete, but the error in the estimate can be as high as 50%. Table 1.2 provides a good example of information typically employed for order-of-magnitude estimates of the capital investment for cell culture facilities. It lists capital investment for cell culture facilities of various sizes built in the last 10 years. The last column displays unit cost of capital investment expressed in millions of US dollars per cubic meter of production bioreactor capacity. The numbers range between 2.5 and 6.2 and for the more recent facilities the numbers are in the 5–6.2 range. Consequently, using the data of Table 1.2, one can safely estimate the capital investment for a new cell culture facility with production bioreactor capacity of 100 m3 to be in the range of $500–650 ...