
eBook - ePub
Proteomics Sample Preparation
Jörg von Hagen, Jörg von Hagen
This is a test
Compartir libro
- English
- ePUB (apto para móviles)
- Disponible en iOS y Android
eBook - ePub
Proteomics Sample Preparation
Jörg von Hagen, Jörg von Hagen
Detalles del libro
Vista previa del libro
Índice
Citas
Información del libro
This long-awaited first guide to sample preparation for proteomics studies overcomes a major bottleneck in this fast growing technique within the molecular life sciences. By addressing the topic from three different angles -- sample, method and aim of the study -- this practical reference has something for every proteomics researcher. Following an introduction to the field, the book looks at sample preparation for specific techniques and applications and finishes with a section on the preparation of sample types. For each method described, a summary of the pros and cons is given, as well as step-by-step protocols adaptable to any specific proteome analysis task.
Preguntas frecuentes
¿Cómo cancelo mi suscripción?
¿Cómo descargo los libros?
Por el momento, todos nuestros libros ePub adaptables a dispositivos móviles se pueden descargar a través de la aplicación. La mayor parte de nuestros PDF también se puede descargar y ya estamos trabajando para que el resto también sea descargable. Obtén más información aquí.
¿En qué se diferencian los planes de precios?
Ambos planes te permiten acceder por completo a la biblioteca y a todas las funciones de Perlego. Las únicas diferencias son el precio y el período de suscripción: con el plan anual ahorrarás en torno a un 30 % en comparación con 12 meses de un plan mensual.
¿Qué es Perlego?
Somos un servicio de suscripción de libros de texto en línea que te permite acceder a toda una biblioteca en línea por menos de lo que cuesta un libro al mes. Con más de un millón de libros sobre más de 1000 categorías, ¡tenemos todo lo que necesitas! Obtén más información aquí.
¿Perlego ofrece la función de texto a voz?
Busca el símbolo de lectura en voz alta en tu próximo libro para ver si puedes escucharlo. La herramienta de lectura en voz alta lee el texto en voz alta por ti, resaltando el texto a medida que se lee. Puedes pausarla, acelerarla y ralentizarla. Obtén más información aquí.
¿Es Proteomics Sample Preparation un PDF/ePUB en línea?
Sí, puedes acceder a Proteomics Sample Preparation de Jörg von Hagen, Jörg von Hagen en formato PDF o ePUB, así como a otros libros populares de Ciencias biológicas y Bioquímica. Tenemos más de un millón de libros disponibles en nuestro catálogo para que explores.
Información
Part I
Perspectives in Proteomics Sample Preparation
1
Introduction
A lot can happen to a protein in the time between its removal from an intact biological system and its introduction into an analytical instrument. Given the increasing sophistication of methods for characterizing many classes of post-translational modification, an increasing variety of protein-modifying processes need to be kept under control if we are to understand what is biology, and what is noise. Hence, the growing importance of sample preparation in proteomics. One might justifiably say that the generation of good samples is half the battle in this field.
Fortunately, proteomics provides us with good methods for studying sample preparation issues. Two-dimensional electrophoresis of plasma, for example, provides a visual protein fingerprint that allows the immediate recognition of sample handling issues such as clotting, platelet breakage, and extended storage at –20 °C (instead of–80 °C). A deeper exploration of plasma using mass spectrometry-based methods provides a more comprehensive picture, though perhaps more difficult to understand.
Unfortunately, despite the power of these methods, we do not know as much about sample quality and sample processing as we need to. The general attitude to these issues in proteomics has been to focus on the standardization of a few obvious variables and hope that the power of the analytical methods allows the sought-for differences between sample groups to shine through. This short-cut approach is likely to be problematic. Not only do the unrecognized effects of sample preparation differences add noise to the background against which the biological signal must be detected, but the sample preparation effects themselves are occasionally confused with biology. Well-informed skeptics correctly suspect that variables as basic as how blood is drawn or stored can generate spurious biomarker signals if the case and control samples are not acquired in exactly the same way. At this point we do not have adequate definitions of what “in exactly the same way” actually means for any given analytical platform.
These problems point to a need to take sample preparation (including initial acquisition through all the steps leading up to analysis) as a mission-critical issue, worthy of time and effort with our best analytical systems. Published data on differences between serum and plasma protein composition, the effect of blood clotting, is interesting but very far from definitive – and in fact specialists in blood coagulation can offer a host of reasons why this process is not easily controllable (and hence not especially reproducible) in a clinical environment. Even a process as widely relied on as tryptic digestion is not really understood in terms of the time course of peptide release or the frequency with which “non-tryptic” peptides are generated – aspects which are critical for quantitative analysis. These and a host of similar issues can be attacked systematically using the tools of proteomics, with the aim of understanding how best to control and standardize sample preparation. In doing so, we will learn much about the tools themselves, and perhaps resolve the paradox surrounding the peptide profiling (originally SELDI) approach: that is, why it seems to be so successful in finding sample differences, but so unsuccessful in finding differences that are reproducible. Perhaps peptide profiling is the most sensitive method for detecting sample preparation artifacts: if it is, then it may be the best tool to support removal of these artifacts and ultimately the best way to classify and select samples for analysis by more robust methods.
Obviously, it is time to take a close look at sample preparation in proteomics. The reader is encouraged to weigh what is known against what is unknown in the following pages, and contemplate what might be done to improve our control over the complex processes entailed in generating the samples that we use.
2
General Aspects of Sample Preparation for Comprehensive Proteome Analysis
2.1 The Need for Standards in Proteomics Sample Preparation
Sample preparation is not the only step - but it is one of the most critical steps - in proteome research. The quality of protein samples is critical to generating accurate and informative data. As proteomic technologies move in the direction of higher throughput, upstream sample preparation becomes a potential bottleneck. Sample capture, transportation, storage, and handling are as critical as extraction and purification procedures. Obtaining homogeneous samples or isolating individual cells from clinical material is imperative, and for this standards are essential. Advances in microfluidic and microarray technologies have further amplified the need for higher-throughput, miniaturized, and automated sample preparation processes.
The need for consistency and standardization in proteomics has limited the success of solutions for proteomics sample preparation. Without effective standards, researchers use divergent methods to investigate their proteins, making it unrealistic to compare their data sets. Until standards emerge, the continual generation of randomized data sets is likely to contribute to the increasing complexity of proteomics research as well as sample preparation.
Proteomics aims to study dynamically changing proteins expressed by a whole organism, specific tissue or cellular compartment under certain conditions. Consequently, two main goals of proteomics research are to: (1) identify proteins derived from complex mixtures extracted from cells; and (2) quantify expression levels of those identified proteins. In recent years mass spectrometry (MS) has become one of the main tools to accomplish these goals by identifying proteins through information derived from tandem mass spectrometry (MS/MS) and measuring protein expression by quantitative MS methods. Recently, these approaches have been successfully applied in many studies, and can be used to identify 500 to 1000 proteins per experiment. Moreover, they can reliably detect and estimate the relative expression (proteins differentially expressed in different conditions) of high- and medium-abundance proteins, and can measure absolute protein expression (quantitation) of single proteins in complex mixtures. The ideal situation would be to catalogue all of the proteins present in a sample and their respective concentrations.However, this level of precision is not yet available because the number and concentration of proteins that can be resolved limits the proteomics methods.
Sample preparation is crucial to obtaining good data – key proteins lost during initial sample preparation can never be restored!
2.2 Introduction: The Challenge of Crude Proteome Sample Analysis
Protein samples used for proteome analysis are retrieved from isolated cells, whole tissues or body fluids. Each of these protein sources is comprised of many more components than the proteins themselves; these non-protein sources include nucleic acids, lipids, sugars, anda wide varietyofsmall molecules. Furthermore, unlike DNA and RNA, both of which are chemically homogeneous, proteins are very heterogeneous andvary widely with respecttotheir size, charge, hydrophobicity, and structure. In addition, many proteins undergo post-translational modification and may have a variety of non-peptide moieties bound to them. This chemical heterogeneity makes isolating proteins extremely challenging. In addition, exogenous reagents, such as salts, buffers and detergents, are usually added to aid in breaking the cells open to release proteins, and otherwise to prepare the sample for investigation. These non-protein components – or “contaminants” – often interfere with proteome analysis.
Thus, a central issue in proteomics is protein sample preparation and the removal of interfering molecules as the quality of any proteome analysis hinges on the quality of the protein sample used.
Successful protein separation is mainly dependent upon decisions that are made regarding sample preparation. Most researchers analyze those proteins present within a whole homogenate where, in theory, all of the proteins present within an organ, cell or body fluid should be present. Realistically, however, highly abundant, proteins such as actin and tubulin or human serum albumin (HSA), proteins of the complement system and antibodies will comprise a significant portion of this homogenate, and the less-abundant regulatory proteins that are most informative regarding cellular response will not be detectable without proper pretreatment and handling.
The tremendous analytical challenge of proteomics arises from the fact that the proteome is a collection of 30 to 80% of gene products expressed at both low levels (10 to 100 copies per cell) and high levels (10 000 to 100 000 copies per cell). These numbers represent a dynamic range of at least six orders of magnitude in living cells, and of 10 to 12 orders of magnitude in body fluids. Especially, the reliable and quantitative comparison of low-abundant proteins in crude extracts of body fluids such as plasma or serum with this large dynamic range in protein expression [2,6] (see Figure 2.1) is the challenge for the post-genomic era [20], but not less challenging with the currently used techniques in whole proteome approaches of living cells.
Figure 2.1 Reference intervals for 70 protein analytes in plasma. Abundance is plotted on a log scale spanning 12 orders of magnitude. Where only an upper limit is quoted, the lower end of the interval line shows an arrowhead. The classical plasma proteins are clustered to the right (high-abundance), the tissue leakage markers (e.g., enzymes and troponins) are clustered in the center, and cytokines are clustered to the left (low-abundance). Hemoglobin is included (far left) for comparison. Data were obtained from the Specialty Laboratories publications (154). Abbreviations: TPA, tissue plasminogen activator; GCSF, granulocyte colony-stimulating factor; TNF, tumor necrosis factor. (Reproduced from Anderson NL, Anderson NG. (2003), The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics, 2(1): 50).
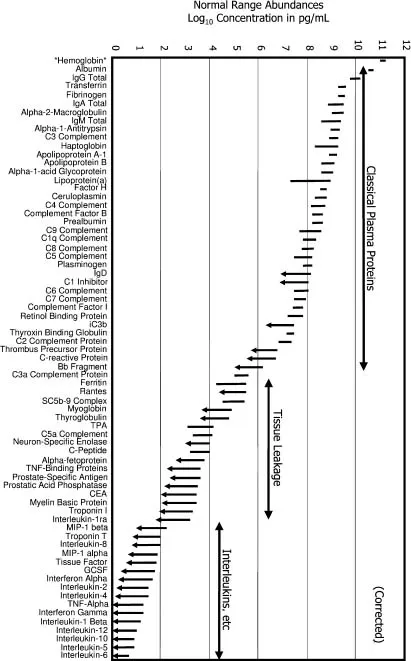
As a point of reference, most eukaryotic cells contain approximately 20000 proteins that have an average molecular weight of 50 kDa. Enzymatic digestion yields approximately 30 peptides per protein, or roughly 6000 000 unique peptides. Certainly, these numbers present at the moment technical challenges in terms of analytical sample throughput, detection and data analysis. The sample preparation techniques of greatest interest in expression proteomics focus on prefractionating and enriching proteins before their separation.
Subcellular fractionation can be conducted as a means to enrich specific orga-nelles and fractions, and thus enable the visualization of significantly more proteins than can be detected in a whole homogenate. However, an increase in workload will result for each additional fraction to be analyzed. Also, sample processing and solubilization will vary for each fraction, and additional troubleshooting is generally needed for optimal reproducibility. Some research groups may be concerned that subcellular fractions prepared from frozen tissue may not be pure due to a loss of membrane integrity during the freeze–thaw process. However, a substantial gain can be realized through the enrichment of subcellular organelles that extends the reach of the researcher in seeking differentially expressed proteins. Nevertheless, researchers should be acutely aware of the need for follow-up studies to validate their assignment of proteins to particular subcel-lular locations or fractions.
This book provides researchers with a practical report of how advanced proteo-mics studies can be used in common laboratory settings by giving a step-by-step description for the sample preparation of the most popular proteomics approaches, and by focusing on reproducible methods and data analysis. We discuss the difficulties and potential sources of variability associated with each phase of proteomics studies, and how these can be addressed. With regard to automated methods, these difficulties include: a high rate of false identifications; the limitations of identifying low-abundance proteins in complex biological samples; the development of analysis methods and statistical models using simple samples; and a lack of validation and a lack of sensitivity and specificity measurements for methods applied to complex samples. Therefore, to maximize protein identification and quantitation and to accelerate biological discovery, it is vital to optimize the methods of sample preparation with regard to the relevant parameters described in the following subsection, as well as separation, data processing and analysis. Finally, we illustrate the utility and value of these methods by describing several diverse proteomics-based studies of diverse species, organisms and up-coming technologies in the field of proteome research.
2.3 General Aspects: Parameters which Influence the Sample Preparation Procedure
Before starting upon your proteomic efforts, its important to spend a few minutes examining the following list, which provides an impression of the parameters which have direct influence on the experimental set-up. Moreover, you might consider additionally any special requisites for your special application, which are not mentioned and described in the following chapters.
- Consider the read-out of the experiment (the aim of the experiment)
- Technically dependent aspects for sample preparation in proteomics:
– The number of samples (single measurements/high-throughput screening)
– Schedule for experimental setting
– Costs per analysis
– Technical equipment in the laboratory (hardware available/sensitivity)
– Use of chemicals/detergents and their effect on down-stream applications and alternatively mechanical disruption methods
– Bioinformatical support (data flow handling)
- Sample-dependent aspects for sample preparation in proteomics:
– Enrichment or depletion strategy: Evaluate the abundance and dynamic range of proteins of interest
– Sample recovery/standardization/storage conditions
– Internal standards (spiking) for quantitative proteome analysis or comparison of diverse samples
– Depending on target protein calculating the amount of sample per analysis (analyte concentration level(s), sample size)
– Developing a procedure which works for different species (Bench to Bedside)
– Sample matrix (origin of sample and isolation of the sample to analyze)
– Localization of target protein (organelle, membrane, cytosolic) has impact on the extraction strategy
Some of these aspects mentioned above, which have a direct impact on the experimental set-up of complex proteome analysis, will be discussed in more detail in the following sections. However, we can begin by drawing some conclusions to the technical aspects of whole-proteome analysis experiments.
2.3.1 Technical Dependent Aspects for Sample Preparation in Proteomics
Before starting the analysisitisimportanttodescribe the aimofthe experiment(s) and thus the number of experiments required for reliable results, including the number of analytes measured intriplicate or,atminimum, duplicate.Ifthe aim isto study effects on signal transduction events (biodiscovery), the number of samples is about 10 to 100 protein samples in sum to discover the proteins involved in a particular signal transduction pathway, or to analyze changes of protein post-translational modifications. If the aim of the test series is to identify a disease-related biomarker, then a significant larger number of samples must be analyzed (up to several thousands) and the identified candidates must be validated under clinical conditions. In both cases, the proteins a priori are not known and thus the approach is labor-intensive, because the requirements for the experimental set-upis to separate inparallel, with a very high resolution, complex protein samples (e.g., a liver lysate with over 3 × 104 different proteins at a given time in a single cell). A further challenge is to characterize the identified protein by sequencing or MS-analysis. The trend in proteome analysis is to quantitate proteins in crude samples; therefore, a set of different labeling strategies is commercially available for MS-based approaches as well as for gel-based methods. These techniques allow the researcher to compare several samples in parallel. Although this often implies a higher accuracy and better reproducibility, and also allows the data to be validated in several independent experiments, it often causes a bottleneck such that the current problem is not efficiently solved. So, based on the considerations described above, the schedule for a basic experiment is from days up to several months or years in biomarker discovery.
If a larger number of protein samples is planned in order to analyze a technique, then a platform selection is needed. Therefore, in addition to the costs per analysis (budget) the numbe of samples per month and the technical equipment availableinthe laboratory mustbeevaluated. Basedon the objective facts with regardtogel separation equipment or MS amenities, the number of expected samples and the sensitivity, the optimized workflow for the proteome analysis must be assigned according to the recommendations described in Figure 2.2. If the appropriate workflow is assigned, the next decision to make is which type of single steps should be combined to answer the question worded in the aim of the experiment. One of the major pit falls with regard to the selected steps is that, often, a simple combination of several single steps is not easily implementable due to the fact that often-applied buffer components such as chemicals/detergen...