![]()
1
Introduction
1.1 WHAT ARE THE TASKS IN PROTEOMICS?
1.1.1 The proteome
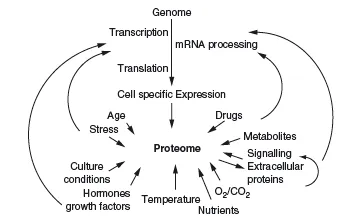
In genomics, one of the main aims is to establish the composition of the genome (i.e. the location and sequence of all genes in a species), including information about commonly seen polymorphisms and mutations. Often this information is compared between different species and local populations. In functional genomics, scientists mainly aim to analyze the expression of genes, and proteomic is even regarded by some as part of functional genomics. In proteomics we aim to analyze the whole proteome in a single experiment or in a set of experiments. We will shortly look at what is meant by the word analysis. Performing any kind of proteomic analysis is quite an ambitious task, since in its most comprehensive definition the proteome consists of all proteins expressed by a certain species. The number of these proteins is related to the number of genes in an organism, but this relation is not direct and there is much more to the proteome than that. This comprehensive definition of the proteome would also account for the fact that not a single individual of a species will express all possible proteins of that species, since the proteins might exist in many different isoforms, with variations and mutations, differentiating individuals. An intriguing example are antibodies, more specifically their antigen binding regions, which exist in millions of different sequences, each created during the lifetime of individuals, without their sequence being predictable by a gene. Antibodies are also a good example of the substantial part played by external influences, which define the proteome; for example, the antibody-mixture present in our bodies is strictly dependent on which antigens we have encountered during our lives. But of course a whole host of more obvious external factors influence our proteome, but not the genome (Figure 1.1).
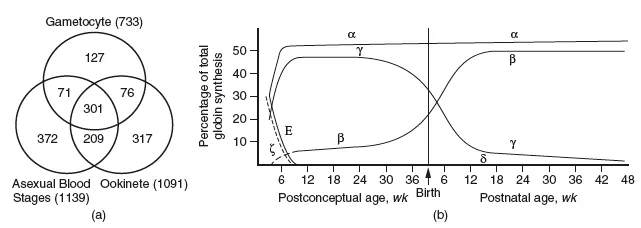
Furthermore, the proteome also contains all possible proteins expressed at all developmental stages of a given species; obvious examples are different proteins in the life cycle of a malaria parasite, or the succession of oxygen binding species during human development, from fetal haemoglobin to adult haemoglobin (Figure 1.2).
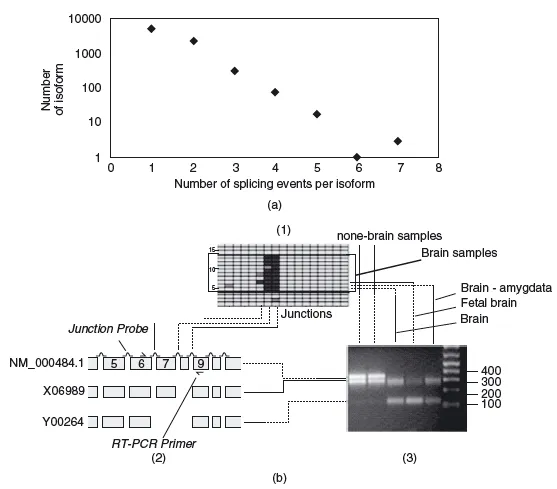
On top of all these considerations, there are possible modifications to the expression of a protein that are not encoded by the sequence of its gene alone; for example, proteins are translated from messenger RNAs, and these mRNAs can be spliced to form different final mRNAs. Splicing is widespread and regulated during the development of every single individual, for example during the maturation of specific cell types. Changes in differential splicing can cause and affect various diseases, such as cancer or Alzheimer's (Figure 1.3).
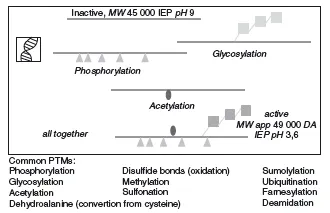
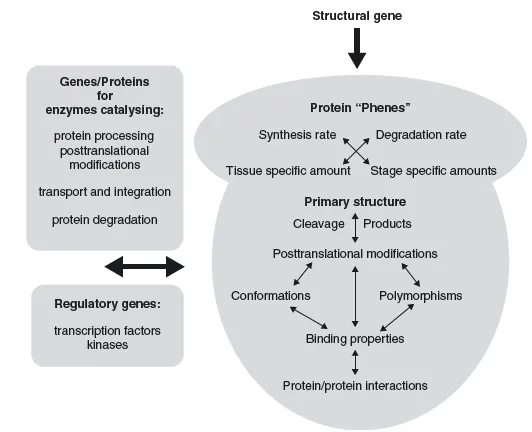
As if all this was not enough variability within the proteome, most proteins show some form of posttranslational modification (PTM). These modifications can be signs of ageing of the protein (e.g. deamidation or oxidation of old cellular proteins; Hipkiss, 2006) or they can be added in an enzymatically regulated fashion after the proteins are translated, and are fundamental to its function. For example, many secreted proteins in multicellular organisms are glycosylated. In the case of human hormones such as erythropoietin this allows them to be functional for longer periods of time (Sinclair and Elliott, 2004). In other cases proteins are modified only temporarily and reversibly, for example by phosphorylation or methylation. This constitutes a very important mechanism of functional regulation, for example during signal transduction, as we will see in more detail later. In summary, there are a host of relevant modifications to proteins that cannot be predicted by the sequence of their genes. These modifications are summarized in Figure 1.4.
Moreover, it is important to remember that the proteome is not strictly defined by the genome. While most possible protein sequences might be predicted by the genome (except antibodies, for example), their expression pattern, PTMs and protein localization are not strictly predictable from the genome. All these factors define a proteome and each protein in it. The genome is the basic foundations for the 'phenotype' of every protein, but intrinsic regulations and external influences also have a strong influence (Figure 1.5).
1.1.2A working definition of the proteome
For all the above mentioned reasons most researchers use a more practical definition of the word 'proteome'; they use it for the proteins expressed in a given organism, tissue/organ (or most likely cell in culture), under a certain, defined condition. These 'proteomes' are then compared with another condition, for example two strains of a microorganism, or cells in culture derived from a healthy or diseased individual. This so-called differential proteomics approach has more than a description of the proteome in mind; its aim is to find out which proteins are involved in specific functions. This is of course hampered by the number of proteins present (some changes may occur as mere coincidences) and by the many parameters that influence the functionality of proteins, expression, modification, localization and interactions. While differential proteomics seems a prudent way to go, we have to keep in mind that the methods chosen for proteomic analyses will also determine the results; for example, if we use a gel-based approach, membrane proteins are almost completely excluded from the analyses. Furthermore, most analyses have a certain cut off level for the low abundant proteins. This means that proteins below (say) 10 000 copies expressed per cell are not easily measurable, because the approaches are usually not sensitive enough.
Even within this limited definition of proteomics we still face substantial tasks, as the proteome is defined not only by the physical state of the proteins in it (expression and modifications) but also by their subcellular location and their membership in protein-protein complexes of ever changing compositions. For instance, it makes a big functional difference to its activity if a transcription factor is inside or outside the nucleus and a proteomic study that fails to analyze the transcription factor's sub-cellular location will miss major changes in the activity of this transcription factor (Figure 1.7). A kinase that needs to be in a multiprotein complex to be active will be inactive when it is only bound to parts of that complex, an important difference that will be missed if we analyze only the presence of a protein but not the interaction partners. The same holds true for kinases that switch complexes and thereby regulate their target specificity (Kolch, 2005).
1.1.3 The tasks in proteomics
Most proteomic studies aim to correlate certain functions with the expression or modification of specific proteins; only few aim to describe complete proteomes or compare them between different species. For a functional correlation we need to analyze the most important protein features of functional relevance. We have already mentioned the analysis of proteins in proteomic studies – just what does this mean? Proteomic analyses can be summarized in terms of specific goals:
1. detection and quantification of protein level;
2. detection and quantification of protein modifications;
3. detection and quantification of sub-cellular protein localization;
4. detection and quantification of protein interactions.
Historically, protein expression has been the first parameter analyzed by proteomics. While this involves a certain form of quantification (present/not present means usually at least a three- to tenfold difference in expression level), it is much harder to quantify proteins on a proteomic scale and many of the latest technological developments focus on this aspect (see Chapters 2–5). Since the abundance of proteins can vary from presumably a single protein to over a million proteins per cell, the quantifications have to cover a dynamic range of over 6 orders of magnitude in cells and up to 10 orders of magnitude in plasma (Patterson and Aebersold, 2003).
PTMs are very important for the function of proteins, and proteomics is the only approach to analyze them on a global scale. Nevertheless, the current approaches (e.g. phosphoproteomics) are by no means able to analyze all possible PTMs, and this remains a hot topic in the development of new technologies.
Before the onset of life cell imaging technology, fractionation of cells was the only method to analyze the subcellular localization of proteins. While being relatively crude and error-prone due to long manipulation times, fractionation studies are very successful in defining protein function. This holds true especially when not only organelles but also functional structures such as ribosomes (Takahashi et al., 2003) or mitotic spindles can be intelligently isolated (Sauer et al., 2005).
The detection of protein interactions is surely the most challenging of proteomic targets, but also a very rewarding one. In single studies the goal is often to identify all interacting partners of a single protein (see Figure 1.8), and several studies taken together can be used to identify, for instance, all interactions within a single signalling module (Bader et al., 2003). Interactions on a truly proteomic scale have been analyzed only in some exceptional studies (Ho et al., 2002; Krogan et al., 2006) and the results are by no means complete, given the temporal and fragile nature of protein-protein interactions, the different results reached with different methods and their complexity.
Non-covalent and hence the most difficult to analyze are localization and interactions of proteins – although none of the above tasks is easily reached, considering the shear number of proteins involved, the minute amounts of sample usually available and the temporal resolution that might be required. Proteomic parameters can change from seconds or minutes (e.g. in signalling) to hours, days and even longer time periods (e.g. in degenerative diseases).
1.2 CHALLENGES IN PROTEOMICS
1.2.1 Each protein is an individual
Nucleotides are made up of four different bases each, and the structure of DNA is usually very uniform. Even if RNA forms more complex structures, we have many different buffers in which we can solubilise all known nucleotides. No such thing exists in proteomics. There is no buffer (and there probably never will be) that can solubilize all proteins of a cell or organism (Figure 1.6). Proteins are made out of 20 amino acids, which allows even a peptide that is 18 amino acids long to acquire more different sequences than there are stars in the galaxy or a hundred times more different sequences than there are grains of sand on our planet!
The average length of proteins is about 450 amino acids. The complexity that can be reached by such a protein is beyond the imagination. More to the point, while almost every sequence of DNA will have fairly similar biochemical properties to any other sequence of similar length, with proteins the situation is totally different. Some proteins will bind to materials used for their extraction and so get lost in analyses, others will appear predominant in a typical mass spectrometry (MS) analysis because they contain optimal amounts and distributions of arginine and lysine. If proteins are very hydrophobic, they will not even get dissolved without the help of detergents. Some proteins show aberrant behaviour with dye; either they are stained easily or very badly. This behaviour makes absolute quantifications and even relative comparisons of protein abundances very difficult. Proteins can display highly dynamic characteristics; their abundances can change dramatically within minutes, by either rapid new synthesis or degradation. Some proteins are more susceptible to degradation by either specific ubiquitin dependent or independent proteolysis than others. ...