No existe en nuestro planeta prácticamente un lugar donde no podamos encontrar bacterias, hallándolas en los sitios más inhóspitos como en la Antártida, en los géiseres de Islandia o en el desierto del Sáhara. Por tanto, están tanto en el exterior como en el interior de los seres vivos.Se estima que en un gramo de arena hay diez millones de bacterias y, solamente, en un mililitro de agua de un río, un millón. Son los seres vivos más numerosos en el planeta, calculándose que hay del orden de cinco quintillones (o cinco billones de trillones), es decir, un trillón de bacterias por cada persona viva.Hay más de 13.000 especies clasificadas, aunque, seguramente, haya muchísimas más sin clasificar, como mínimo otras 30.000, por lo que conocemos una parte muy limitada del mundo bacteriano. Afortunadamente, el 99, 994% de las clasificadas hasta la fecha son inocuas para el hombre; es más, muchas son imprescindibles, ya que son responsables del mantenimiento de los ciclos biogeológicos. Además, han intervenido desde siempre en nuestra dieta, produciendo alimentos y bebidas fermentadas, sirva como ejemplo el yogur, o los productos encurtidos. Nuestra microbiota experimenta cambios, como consecuencia de la influencia de múltiples factores, de un modo similar a los que experimenta cualquier órgano de nuestro cuerpo desde la ontogenia a la muerte. Estos cambios pueden ocurrir en cuestión de días, como ocurre durante la ingesta de antibióticos, o a más largo plazo durante la exposición continuada a una dieta.A pesar de ello, el estudio de la microbiota ha permanecido bastante estancado durante la mayor parte del siglo XX debido a que no se habían desarrollado tecnologías que permitieran el análisis adecuado de las complejas comunidades microbianas que habitan en nuestro organismo y de la enorme variedad de interacciones que se producen. Este conocimiento ha cambiado radicalmente en los últimos años con la caracterización del microbioma humano, que supusieron un hito en la historia de la biomedicina.Las Técnicas Ómicas aparecen para contribuir al estudio de la totalidad de microbios existentes en los seres vivos. A este conjunto de microorganismos se les denomina microbiota y los podemos encontrar en el aparato digestivo, en el reproductor, en el respiratorio, en la boca, en la piel…, en definitiva, sobre todo en las zonas de mayor humedad del organismo. La relación de simbiosis entre humano y los miembros de la microbiota es el resultado evolutivo de una interacción biológica en la que, normalmente, una o ambas partes obtienen beneficio.Ahora sabemos que el estilo de vida actual ha ejercido un fuerte impacto en nuestra microbiota y que algunos de los microorganismos ancestrales y los genes que estos contienen han ido perdiéndose o disminuyendo.

eBook - ePub
Técnicas ómicas aplicadas al estudio de la microbiota
- 150 páginas
- Spanish
- ePUB (apto para móviles)
- Disponible en iOS y Android
eBook - ePub
Técnicas ómicas aplicadas al estudio de la microbiota
Descripción del libro
Cuenta con la confianza de 375,005 estudiantes
Acceso a más de 1,5 millones de títulos por un precio mensual justo.
Estudia de forma más eficiente usando nuestras herramientas de estudio.
Información
Categoría
MedicineCategoría
Genetics in MedicineCapítulo 1
Tecnologías de secuenciación masiva
Llucia Martínez Priego, Giuseppe D’Auria
Las descripciones de las poblaciones microbianas de ambientes naturales, así como aquellas relacionadas con organismos superiores, pasaron a finales del siglo pasado por un punto de inflexión debido al desarrollo de las tecnologías de secuenciación masiva. El impulso que la secuenciación masiva ha proporcionado a la biología molecular procede fundamentalmente de la eliminación de los pasos de clonaje acelerando los procesos y abaratando los costes por base secuenciada. Para entender el avance que ha supuesto hay que recordar que los primeros trabajos de metagenómica estuvieron basados en la extracción directa del ADN total de una comunidad microbiana, fragmentándolo e insertándolo en plásmidos o BACs (del inglés, Bacterial Artificial Chromosome) de bacterias de laboratorio (generalmente Escherichia coli), obviando así la necesidad de cultivo. Los clones obtenidos podrían entonces utilizarse para la secuenciación de tipo Sanger y posterior estudio funcional1. Este proceso se llevaba a cabo en serie (un clon tras otro) y aplicando la fuerza bruta, es decir, secuenciando el número suficiente de clones y de fragmentos de ADN que permitiera describir los organismos. El salto que supuso poder estudiar también los organismos no cultivables permitió entender mejor y describir cómo los organismos no viven aislados, sino en continua interacción con comunidades microbiológicas complejas en prácticamente cualquier ambiente.
Tras este primer avance, en la primera década de este nuevo siglo, aparece por primera vez una nueva generación de secuenciadores llamados secuenciadores «masivos» o secuenciadores de «segunda generación» (la secuenciación Sanger se considera la «primera generación»). Estos secuenciadores permitieron pasar de la secuenciación en serie (clon por clon) a la secuenciación en paralelo de cientos de miles de fragmentos de ADN en una sola reacción, evitando el costoso paso del clonaje y abaratando los costes y los tiempos. Estos avances, junto con la reducción de la cantidad de ADN requerido para la construcción de las librerías, ha posibilitado la expansión de los estudios metagenómicos así como la creación de consorcios locales o internacionales destinados a analizar y comprender la diversidad génica y poblacional de los distintos microbiomas en prácticamente todos los ambientes conocidos. También se ha disparado el número de proyectos de metataxonomía basados en la descripción de las distribuciones de amplicones del gen ribosomal 16S, conocido como la etiqueta taxonómica más importante por su estabilidad evolutiva2.
Es importante también señalar cómo estos avances han ido de la mano con otros procedentes del mundo de la informática, donde procesadores cada vez más potentes han hecho posible el almacenamiento y análisis de esta cantidad masiva y creciente de datos, dando origen a una nueva disciplina, la bioinformática. En su comienzo asistimos a un avance muy «creativo», dando tumbos según las capacidades y la inventiva de los biólogos que se acercaban a la biología computacional confluyendo finalmente en la nueva figura del bioinformático. Finalmente, nuevos estándares en términos de calidad, descripción de los datos y métodos de análisis empiezan a afirmarse para contener el caos inicialmente generado por tanta información, llegada en tan poco tiempo a la portada de todos los grupos de investigación.
1.1 Dónde hemos llegado: tres generaciones de secuenciadores
-Primera generación. Los primeros abordajes a la secuenciación masiva empiezan con la aparición de instrumentos automatizados de secuenciación de tipo Sanger basados en el análisis seriado de fragmentos de ADN clonados. El número de fragmentos que se podían obtener por unidad de tiempo era dependiente del número de capilares del secuenciador, donde cada uno de ellos realizaba una electroforesis individual. Las lecturas así obtenidas estaban caracterizadas por una alta calidad (medida como la probabilidad inversa de que la base no fuera correcta), con una longitud de hasta 900 pb. Las secuencias se visualizaban y comprobaban de una en una pudiendo generar cientos de secuencias al día por un coste de unos pocos euros por secuencia.
-Segunda generación. Empieza la secuenciación masiva o en paralelo. Estas tecnologías tienen en común la fragmentación de los genomas y la amplificación clonal de los fragmentos obtenidos. Finalmente, mediante la inclusión de los clones en gotas de aceite (emulsion PCR) o su inmovilización en una superficie sólida, consiguen amplificar la señal necesaria para detectar la secuencia de las bases nucleotídicas. La diferencia entre los secuenciadores y las diferentes tecnologías radica en la diferente química que aplican para detectar la señal. La primera aproximación a la secuenciación masiva en paralelo se basó en la pirosecuenciación del ADN3. Esta tecnología fue desarrollada por la empresa 454 Life Sciences, que fue absorbida después por la empresa Roche. El último modelo que comercializó en 2010, el GS-FLX-Titanium+ generaba lecturas de hasta 800 bases al ritmo de 100 millones de bases cada 4 horas. Otras compañías desarrollaron otro tipo de tecnologías: la compañía Solexa (desde 2007 illumina) desarrolló un método basado en la polimerización del ADN y la compañía SOLiD (Sequencing by oligonucleotide ligation and detection), un método basado en la detección de fluorescencia por ligación de sondas. Ambas técnicas tienen la gran ventaja sobre la pirosecuenciación de resolver de forma fiable las regiones homopoliméricas (repeticiones de las mismas bases), sin embargo, su gran desventaja radica en que no son capaces de generar lecturas largas. Otra compañía, IonTorren Systems Inc. (en la actualidad, comercializado por ThermoFisher) desarrolló un método basado en la detección de variaciones de pH durante la polimerización. Actualmente, el máximo de longitud lo alcanzan los secuenciadores de tipo MiSeq de illumina con secuencias emparejadas de 2x300 pb, es decir, no más de 600 pb frente a los 800 pb que tenía la pirosecuenciación (y los 900 pb con los que se podía llegar con el método de Sanger de primera generación).
-Tercera generación. La nanotecnología se aunó con la biología para desarrollar nuevas tecnologías de secuenciación que actúan a nivel molecular, conocidos como secuenciadores de tercera generación se basan en la secuenciación de una única molécula de ADN sin necesidad de amplificar clonalmente el ADN y evitando de esta manera introducir sesgos relacionados con esa amplificación. Actualmente, dos compañías lideran esta tercera generación, Oxford Nanopore, con su tecnología de nanoporos por los que pasan las bases de ADN produciendo diferentes alteraciones de potencial eléctrico que las identifica, y Pacific Biosciences, con su tecnología de SMRT (del inglés, single molecule real time sequencing) y nanopocillos en los que se individualizan y se leen con precisión las moléculas de ADN. Aunque Oxford Nanopore es, a fecha de hoy, mejorable en términos de calidad de las lecturas y Pacific Bioscience, en términos de cantidad de ADN requerido y precio de la secuenciación, ambas representan un paso enorme en los estudios de genómica, metagenómica y descripciones poblacionales bacterianas y virales4.
1.2 Genomas completos: desde la fuerza bruta a la elegancia
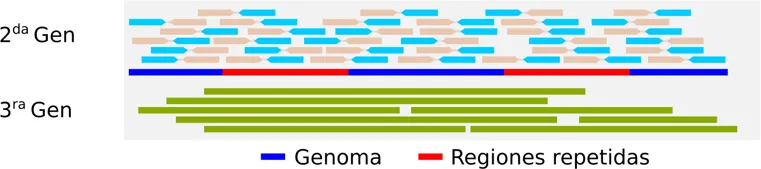
La secuenciación de genomas completos empleando secuenciación de primera generación ha sido un trabajo muy largo y costoso. Cientos de secuencias de Sanger, librería tras librería, procedentes de la clonación de fragmentos de ADN en plásmidos, fósmidos y PCR inversas para intentar cerrar agujeros o resolver regiones particularmente complicadas. Siempre, el mayor quebradero de cabeza en cada proyecto genoma ha sido representado por la resolución de las regiones repetidas, ya que los métodos de secuenciación de primera y segunda generación, muchas veces, no permiten superar las extensiones de dichas repeticiones (secuencias de inserción, transposones o los mismos operones ribosomales), produciendo fragmentos de ensamblados imposibles de colocar en el correcto orden en el genoma. Aunque las tecnologías han ido aumentando el número de lecturas obtenidas, es decir, la cobertura del genoma, en estos casos no ha sido de ayuda, ya que aumentar la cobertura no resuelve el problema de conseguir saltar por encima de las repeticiones. El aumento de cobertura solamente ayuda en la corrección de errores debidos a baja calidad de secuenciación o en afinar algunas regiones homopolimericas. Para paliar este problema se han desarrollado varios protocolos de creación de librerías conocidos como mate pairs, los cuales, mediante la circularización y etiquetado de fragmentos largos, permiten secuenciar los extremos de esos fragmentos separados por kilobases, guiando así los ensambladores en la ordenación de los contigs (secuencias consenso de secuencias solapantes) y cerrando los genomas de manera más ágil y exacta. Finalmente, el gran éxito de la segunda generación ha sido sin lugar a dudas el aumento de la cantidad de lecturas obtenidas en una carrera y, por tanto, la reducción del coste de secuenciación por genoma. La tercera generación de secuenciadores, al aumentar la longitud de las secuencias a decenas de kilobases, facilita enormemente el cierre de los genomas superando el problema de las regiones repetidas (Figura 1). Ahora es ya una realidad obtener en una sola carrera (o fracción de ella) el genoma completo de un organismo sin necesidad de pasar por un tedioso proceso de acabado manual5.
El salto de estas tecnologías ha sido tan importante que el coste por genoma ha pasado de cientos de miles de euros y años de trabajo a pocos días (2 o 3) para obtener las secuencias y pocas horas para ensamblar y anotar por tan solo unos cientos de euros por muestra. Lo que se hipotetizaba como genómica en tiempo real, es hoy una realidad6. Se puede secuenciar y conocer la genética de un organismo patógeno o la evolución o estabilidad de un probiótico en tiempos inferiores a los 60 minutos, y trabajando in situ, o sea, en el mismo sitio donde se encuentra el organismo de interés sin necesidad de laboratorios completos4.
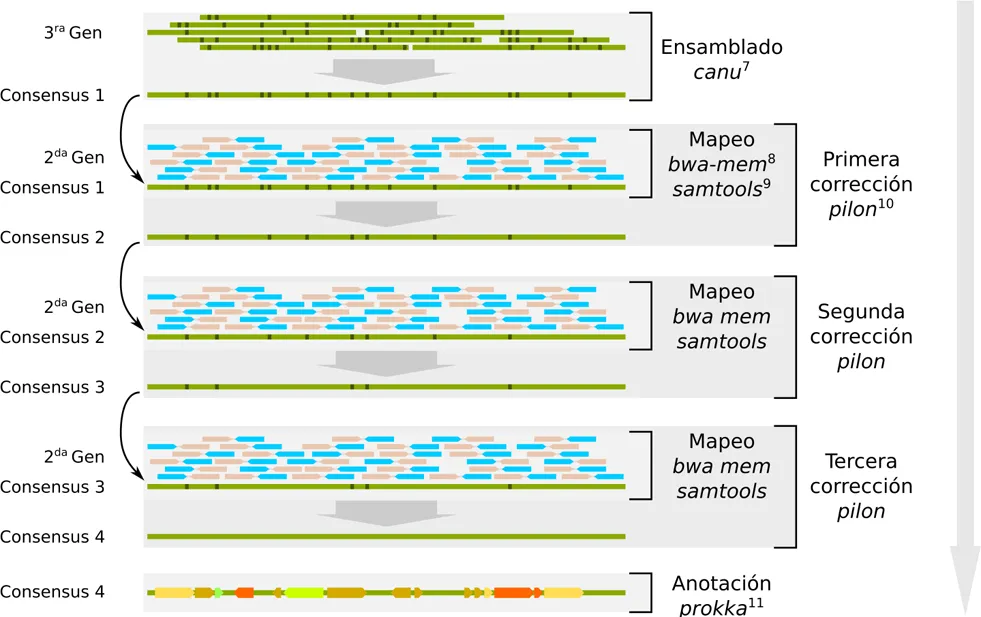
En concreto, hoy en día el mejor abordaje a la genómica de novo consiste en llevar a cabo ensamblados híbridos entre secuencias largas y cortas (tercera y segunda generación). La primera permite llevar a cabo el proceso de scaffolding, es decir, construir el primer andamio del genoma; las secuencias largas permiten superar las repeticiones acabando el genoma teóricamente (y muchas veces prácticamente) en una sola pieza que resembla el genoma original y, de la misma manera, se obtienen en un solo experimento también elementos extracromosómicos como los plásmidos asociados al genoma de interés. Se puede llegar a este punto con un esfuerzo de secuenciación de alrededor 20-40x de cobertura. Ahora bien, sabemos que de momento los secuenciadores de tercera generación tienen todavía un nivel de errores considerable, que podemos corregir con secuencias de segunda generación (con una cobertura adicional de ~100x), cuyo nivel de errores es relativamente bajo. Existen ya programas bioinformáticos que permiten corregir estos errores empleando el ensamblado de tercera generación como base y las secuencias de segunda generación para corregir los errores. El protocolo se puede resumir en el flujo de trabajo (pipeline) expresado en la Figura 2. Al final del proceso nos encontraremos con un genoma completo y anotado.
Entre los secuenciadores de tercera generación, Oxford Nanopore aparece en el mercado, entre otras configuraciones, con el secuenciador MinIon, del tamaño de un pen-drive, óptimo para un uso realmente portátil conectado directamente a un ordenador también portátil vía USB. Este secuenciador tiene los tiempos más cortos de preparación de librerías (una hora) y se caracteriza por la producción de datos de secuencias en tiempo real. Por primera vez, podemos llevar a cabo un análisis, por ejemplo, de tipo diagnóstico, directamente in situ. El número de escenarios de aplicaciones se dispara: desde la búsqueda de agentes infecciosos in situ hasta el control de dinámicas de poblaciones en fermentadores en tiempo casi real.
La secuenciación de genomas bacterianos completos tiene ahora un frente de investigación en la epidemiología molecular, dado que tanto el tiempo de generación de datos de secuencias como el coste son muy bajos (~ 150 euros). Por esta razón es ya posible seguir los brotes o infecciones nosocomiales usando los perfiles de mutaciones y para evaluar distancias evolutivas entre los aislados y la cepa de referencia para las cuales ya se tiene su genoma completo. Este abordaje tien...
Índice
- Cubierta
- Créditos
- Sumario
- Autores
- Prólogo
- Capítulo 1: Tecnologías de secuenciación masiva
- Capítulo 2: Del gen a la función: metatranscriptómica, metaproteómica y metabolómica microbiana
- Capítulo 3: Herramientas bioinformáticas
- Capítulo 4: El efecto del genoma del huésped en el microbioma intestinal
- Capítulo 5: La metabolómica: una herramienta esencial en el estudio del metabolismo y la microbiota
- Capítulo 6: Integración de técnicas ómicas en el estudio de la microbiota intestinal
- Capítulo 7: Microbiota mamaria
- Capítulo 8: Ginómica (microbiota vaginal)
Preguntas frecuentes
Sí, puedes cancelar tu suscripción en cualquier momento desde la pestaña Suscripción en los ajustes de tu cuenta en el sitio web de Perlego. La suscripción seguirá activa hasta que finalice el periodo de facturación actual. Descubre cómo cancelar tu suscripción
No, los libros no se pueden descargar como archivos externos, como los PDF, para usarlos fuera de Perlego. Sin embargo, puedes descargarlos en la aplicación de Perlego para leerlos sin conexión en el móvil o en una tableta. Descubre cómo descargar libros para leer sin conexión
Somos un servicio de suscripción de libros de texto en línea, donde puedes acceder a toda una biblioteca digital por menos del precio de un solo libro al mes. Con más de 1,5 millones de libros en más de 990 categorías, ¡te tenemos cubierto! Conoce nuestra misión
Busca el símbolo de lectura en voz alta en tu próximo libro para ver si puedes escucharlo. La herramienta de lectura en voz alta lee el texto en voz alta por ti, resaltando el texto a medida que se lee. Puedes pausarla, acelerarla y ralentizarla. Obtén más información sobre la lectura en voz alta
¡Sí! Puedes usar la aplicación de Perlego en dispositivos iOS y Android para leer cuando y donde quieras, incluso sin conexión. Es ideal para cuando vas de un lado a otro o quieres acceder al contenido sobre la marcha.
Ten en cuenta que no será compatible con los dispositivos que se ejecuten en iOS 13 y Android 7 o en versiones anteriores. Obtén más información sobre cómo usar la aplicación
Ten en cuenta que no será compatible con los dispositivos que se ejecuten en iOS 13 y Android 7 o en versiones anteriores. Obtén más información sobre cómo usar la aplicación
Sí, puedes acceder a Técnicas ómicas aplicadas al estudio de la microbiota de Abelardo Margolles Barros,Rafael Bargiela Bargiela,Aitor Blanco-Míguez,Xavier Correig Blanchar,Mauro D'Amato,Giuseppe D'Auria,Manuel Ferrer Martínez,Koldo García Etxebarria,Llucia Martínez Priego,Celia Méndez-García,Andrés Moya Simarro,Vicente Pérez Brocal,Juan Miguel Rodríguez Gómez,Lorena Ruiz Gar en formato PDF o ePub, así como a otros libros populares en Medicine y Genetics in Medicine. Tenemos más de 1,5 millones de libros disponibles en nuestro catálogo para que explores.