Negli ultimi 10 anni si sono registrati progressi molto significativi nello sviluppo di piccole molecole per il trattamento della fibrosi cistica (CF), molecole che sono correttori o potenziatori del canale CFTR. Tuttavia, queste molecole incrementano rispettivamente la quantità del canale nella membrana plasmatica e la probabilità di apertura del canale. Correttori e potenziatori per agire richiedono quindi la presenza della proteina
Tuttavia, circa il 10% della popolazione affetta da CF mostra mutazioni non-senso che causano la formazione di un codone di terminazione prematura (PTC) associato a una proteina CFTR incompleta e non funzionale. In questo contesto genetico i correttori e i potentatori non possono agire.
Una potenziale terapia per mutazioni non-senso è quella di migliorareil readthrough del PTC mediante l'uso di piccole molecole, denominate Translational Readthrough Inducing Drugs (TRIDs). Al fine di testare questi farmaci, abbiamo prodotto un modello CF utilizzando cellule tiroidee di ratto (FRT) che non esprimono il gene CFTR endogeno.
In queste cellule, abbiamo trasfettato un vettore plasmidico contenente la mutazione CFTR-W1282X (UGA-opale) che è stato usato per testare tre nuove molecole di TRIDs.

eBook - ePub
Produzione di cellule FRT CFTR-W1282X per lo studio avanzato di molecole ad azione readthrough (TRIDs)
- Italian
- ePUB (disponibile su mobile)
- Disponibile su iOS e Android
eBook - ePub
Produzione di cellule FRT CFTR-W1282X per lo studio avanzato di molecole ad azione readthrough (TRIDs)
Informazioni su questo libro
Scelto da 375,005 studenti
Accedi a oltre 1 milione di titoli a un prezzo mensile contenuto.
Studia in modo più efficiente con i nostri strumenti dedicati.
Informazioni
Argomento
Biological SciencesCategoria
Molecular BiologyIntroduzione
La Fibrosi Cistica
Con circa 70.000 persone affette e 1000 nuovi casi l’anno, la Fibrosi Cistica (FC) è la malattia genetica letale più diffusa al mondo, con una mortalità che si attesta a circa il 90% dei casi [Misbahuddin M, 2017]. Un individuo su venticinque è portatore sano di questa patologia monogenica con ereditarietà autosomica recessiva, le cui manifestazioni cliniche sono causate da mutazioni del gene CFTR (Cystic Fibrosis Transmembrane Conductance Regulator).
L’omonima proteina, che da questo gene viene codificata, è un canale anionico regolato dall’cAMP, espresso sulla superficie apicale delle cellule epiteliali di differenti tessuti e organi secretori (polmoni, pancreas, intestino, genitali), dove consente il passaggio dello ione cloro, seguito per osmosi dal flusso di acqua, che rende quindi fluido il muco che riveste i suddetti tessuti.
Sono state identificate quasi duemila mutazioni del gene CFTR che determinano alterazioni strutturali e/o funzionali dell’omonimo canale e che causano quindi un ridotto trasporto dello ione cloro, predisponendo così, ad un ispessimento dello strato di secrezione mucosa con conseguente ostruzione e danno tissutale [Bradley S, 2016].
Le manifestazioni cliniche più frequenti di questa patologia multiorgano, sono infatti, ritardo nella crescita, causato da una ridotta secrezione di enzimi pancreatici e conseguente difficoltà nell’assorbimento di grassi e proteine, e alterazioni respiratorie che frequentemente evolvono in infezioni polmonari croniche [Castellani C, 2017].
Lo spesso strato di muco risulta essere infatti un ambiente ottimale per la proliferazione batterica, che determina uno stato di infiammazione cronica a cui segue un massivo intervento di neutrofili [Misbahuddin M, 2017].
Il meccanismo d’azione di questi leucociti, nel tentativo di ridurre l’infezione, causa tuttavia il rilascio di un’elevata concentrazione di acidi nucleici e proteine della matrice, che contribuiscono insieme al rilascio di elastasi e citochine proinfiammatorie, all’incremento della viscosità del muco e al danno tissutale [Misbahuddin M, 2017].
Le terapie per la FC, prima rivolte esclusivamente alla riduzione della sintomatologia, si stanno adesso sempre più rivolgendo alla causa fondante della patologia, nel tentativo di individuare una terapia univoca che sia in grado di ripristinare la funzionalità del canale CFTR e che sia valida per la grande varietà di mutazioni che colpiscono questo gene.
Il gene CFTR e le sue mutazioni
Il gene CFTR, lungo circa 230 kb, è localizzato sul braccio lungo del cromosoma 7 e codifica per una proteina di 1480 amminoacidi; al termine della traduzione, essendo una proteina di membrana, CFTR attraverserà reticolo endoplasmatico e apparato del Golgi, andando incontro a complesse modifiche post-traduzionali che le consentiranno di raggiungere il corretto ripiegamento. Le molteplici mutazioni a carico di questo gene possono essere distinte in sei classi in base all’alterazione causata e alla gravità delle manifestazioni cliniche [Iwona Pranke et al. 2019 ].
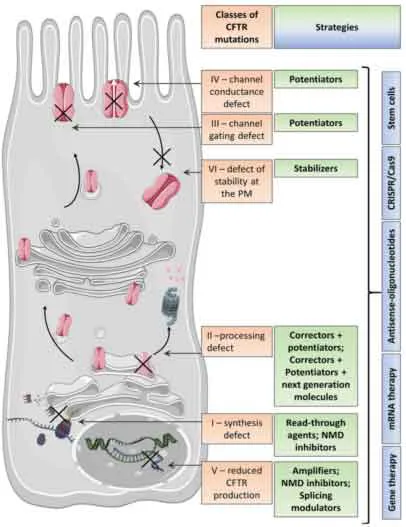
Figura. 1: Classificazione delle mutazioni a carico di CFTR e corrispondenti strategie terapeutiche [Iwona Pranke et al. 2019].

Le differenti classi determinano manifestazioni fenotipiche ampiamente eterogenee sia nel grado di severità della patologia, sia nella velocità di progressione della stessa, con le forme più severe che si identificano nelle classi I, II e III.
Una maggiore variabilità è determinata anche dalle diverse combinazioni alleliche in cui le mutazioni possono trovarsi a livello genomico.
Riveste, quindi, sempre più importanza la genotipizzazione, non solo per conferma della diagnosi, ma anche per l’identificazione del trattamento più appropriato e la personalizzazione della terapia [Ramsey et al. 2011].
Le manifestazioni fenotipiche della patologia sono però significativamente differenti anche in pazienti presentanti le stesse mutazioni, con un decorso non sempre prevedibile sulla base degli esami clinici e molecolari. Sul fenotipo finale influiscono infatti fattori ambientali e variazioni geniche che riguardano locus diversi da quello di CFTR. Ne sono esempi, i polimorfismi a carico dei geni coinvolti nel pathway NMD o del gene codificante per il fattore di crescita trasformante β (TGFβ). [Drumm et al. 2005]. Per queste ragioni, l’identificazione di una terapia univoca è un obiettivo complesso da raggiungere. Nonostante ciò, alla terapia genica, vanno progressivamente aggiungendosi trattamenti con piccole molecole capaci di agire sul difetto proteico o al livello del trascritto di CFTR.
Struttura della proteina CFTR
CFTR è una proteina canale facente parte della superfamiglia delle ATP-binding cassette (ABC). Nonostante condivida con le proteine ABC le principali caratteristiche strutturali, La proteina CFTR possiede proprietà funzionali uniche che lo rendono essenziale per il mantenimento della fisiologia dei tessuti in cui è espresso.
Gli altri membri di questa superfamiglia sfruttano infatti l’idrolisi dell’ATP per determinare un trasporto attivo dei rispettivi substrati, al contrario di CFTR in cui il passaggio degli ioni avviene secondo gradiente elettrochimico e il legame dell’ATP è funzionale solo all’apertura del canale [Theodoulou and Kerr, 2015].
CFTR è un canale relativamente non selettivo, permeabile agli anioni monovalenti grazie alla presenza, sulla superficie del poro di permeazione, di residui di arginine e lisine, che, con la loro carica positiva, attraggono gli anioni e respingono i cationi [Fangyu Liu et al., 2017]. Il poro è definito da due domini transmembrana (TMD1, TMD2), ciascuno formato da 6 α-eliche che si connettono alla componente citoplasmatica del canale, composta da due domini NBD (nucleotide-binding domain), con il ruolo di legare l’ATP. Secondo il meccanismo di apertura del canale, l’idrolisi dell’ATP avverrà soltanto quando i due NBDs formeranno un dimero, che consentirà c...
Indice dei contenuti
- Cover
- Indice
- Frontespizio
- INTRODUZIONE
- SCOPO DELLA RICERCA
- MATERIALI E METODI
- RISULTATI
- DISCUSSIONE
- BIBLIOGRAFIA
- SITOGRAFIA
- RINGRAZIAMENTI
Domande frequenti
Sì, puoi annullare l'abbonamento in qualsiasi momento dalla sezione Abbonamento nelle impostazioni del tuo account sul sito web di Perlego. L'abbonamento rimarrà attivo fino alla fine del periodo di fatturazione in corso. Scopri come annullare l'abbonamento
No, i libri non possono essere scaricati come file esterni, ad esempio in formato PDF, per essere utilizzati al di fuori di Perlego. Tuttavia, puoi scaricarli nell'app Perlego per leggerli offline su smartphone o tablet. Scopri come scaricare libri offline
Perlego offre due piani: Essential e Complete
- Essential è l'ideale per studenti e professionisti che amano esplorare un'ampia gamma di argomenti. Accedi alla libreria Essential, che include oltre 800.000 titoli di comprovata qualità e bestseller in vari settori, tra cui business, crescita personale e discipline umanistiche. Include tempo di lettura illimitato e voce standard per la sintesi vocale.
- Complete: perfetto per studenti e ricercatori esperti che necessitano di un accesso completo e illimitato. Accedi a oltre 1,4 milioni di libri su centinaia di argomenti, inclusi titoli accademici e specialistici. Il piano Complete include anche funzionalità avanzate come la sintesi vocale premium e l'assistente di ricerca.
Perlego è un servizio di abbonamento a testi accademici, che ti permette di accedere a un'intera libreria online a un prezzo inferiore rispetto a quello che pagheresti per acquistare un singolo libro al mese. Con oltre 1 milione di testi suddivisi in più di 990 categorie, troverai sicuramente ciò che fa per te! Scopri la nostra missione
Cerca l'icona Sintesi vocale nel prossimo libro che leggerai per verificare se è possibile riprodurre l'audio. Questo strumento permette di leggere il testo a voce alta, evidenziandolo man mano che la lettura procede. Puoi aumentare o diminuire la velocità della sintesi vocale, oppure sospendere la riproduzione. Scopri di più sulla funzione di sintesi vocale
Sì! Puoi utilizzare l'app di Perlego su dispositivi iOS e Android per leggere quando e dove vuoi, anche offline. È perfetta per gli spostamenti quotidiani o quando sei in viaggio.
I dispositivi con iOS 13 e Android 7 o versioni precedenti non sono supportati. Scopri di più su come utilizzare l'app
I dispositivi con iOS 13 e Android 7 o versioni precedenti non sono supportati. Scopri di più su come utilizzare l'app
Sì, puoi accedere a Produzione di cellule FRT CFTR-W1282X per lo studio avanzato di molecole ad azione readthrough (TRIDs) di Roberta Bongiorno in formato PDF e/o ePub, così come ad altri libri molto apprezzati nelle sezioni relative a Biological Sciences e Molecular Biology. Scopri oltre 1 milione di libri disponibili nel nostro catalogo.