![]()
Chapter 1
______________________
Introduction
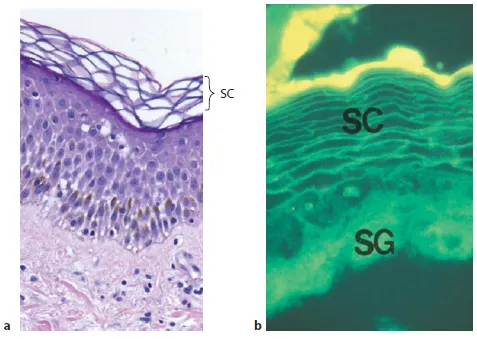
Generalized scaling disorders can be of either acquired or inherited etiology. This book focuses solely on generalized, inherited (mendelian) disorders of cornification (DOC or MeDOC), which constitute an ever-enlarging group of monogenic diseases caused by a large number of genes that affect a broad array of cellular functions (table 1). The diagnosis of specific entities within this group largely rests upon recognition of specific clinical features (e.g. the quality and distribution of scales, the neonatal phenotype and the presence or absence of associated cutaneous abnormalities, such as ectropion, keratoderma, centripetal vs. acral involvement, hair shaft anomalies) as well as involvement of other organ systems. With the exception of the characteristic light-microscopic features of epidermolytic ichthyosis (EI; epidermolytic hyperkeratosis, EHK), the droplets positive for oil red O in neutral lipid storage disease and ichthyosis prematurity syndrome (IPS; and the characteristic lamellar inclusions in the corneocyte cytosol in IPS), routine histopathology and ultrastructure do not suffice to allow the correct diagnoses. There are several reasons for this. First, images of the stratum corneum (SC), as viewed by light as well as by routine electron microscopy, are largely artifactual in appearance. For example, because of shrinkage and extraction of extracellular lipids during routine tissue processing, the ‘normal basket weave pattern’ of the SC in no way reflects the true architecture of this tissue (fig. 1a). If parallel samples instead are viewed as frozen sections (where lipid extraction is avoided) and stained with lipophilic dyes, both the compact, cohesive, organized structure of normal SC, and the localization of lipids to intercellular membrane domains can be appreciated (fig. 1b).
The second reason for the limited utility of light microscopy in the diagnosis of the ichthyoses is perhaps even more important, namely the convergence of a multiplicity of genotypes upon a limited spectrum of clinical phenotypes. This phenotypic convergence can be best understood by consideration of the impact of the mutations on SC function, particularly permeability barrier function, and the homeostatic mechanisms that are activated in an attempt to correct barrier dysfunction - efforts that are at best only partially successful. The metabolic response to barrier failure includes: (1) upregulation of lipid synthesis in nucleated epidermal cell layers and accelerated delivery of more lipids to the SC (the ‘make and deliver more lipid!’ imperative); (2) epidermal hyperproliferation (the imperative to ‘make more cells that in turn will make more lipid’), and (3) inflammation (‘protect from invading microorganisms!’).
Table 1. Functional classification of the ichthyoses
Category | Disorders |
Protease/ antiprotease | Netherton syndrome, Papillon-Lefèvre syndrome |
Lipid metabolism | Refsum disease, neutral lipid storage disease with ichthyosis, Sjögren-Larsson syndrome, congenital hemidysplasia with ichthyosiform erythrodermaand limb defects, Conradi-Hünermann-Happle syndrome, recessive X-linked ichthyosis, Gaucher disease |
Lipid assembly/ transport | Harlequin ichthyosis, cerebral dysgenesis/neuropathy/ ichthyosis/palmar-plantar keratoderma syndrome, mental retardation/enteropathy/deafness/neuropathy/ ichthyosis/keratoderma syndrome, ichthyosis prematurity syndrome |
Keratinopathies | Epidermolytic ichthyosis, superficial epidermolytic ichthyosis |
Corneocyte envelope | Loricrin keratoderma, transglutaminase-1-negative lamellar ichthyosis |
DNA transcription | Trichothiodystrophy |
Cell-to-cell communication | Erythrokeratoderma variabilis, Vohwinkel syndrome (connexins) |
Thus, the net consequences of epidermal hyperplasia, hyperkeratosis and inflammation are near-universal features of the ichthyoses. The interplay of this limited array of repair responses confronting the flawed cellular consequences of the specific genotype results in the specific, albeit often overlapping, clinical phenotypes.
Attempts have been made to utilize the higher resolution offered by routine electron microscopy to refine diagnoses of this heterogeneous group of inherited disorders, but an important limitation of routine electron microscopy is that standard techniques do not permit evaluation of either the quantity or the organization of the lipid-enriched, extracellular matrix of the SC. Standard processing of tissue samples for electron microscopy results in the same extraction artifacts that occur during paraffin embedding for light microscopy. Hence, key information about abnormalities in the extracellular compartment of the SC cannot be retrieved. The limited progress to date in delineating the pathogenesis of many of the DOC can be attributed largely to a failure to utilize methods that allow evaluation of dynamic changes in the architecture of affected SC, including not only changes in the organization of the lipid-enriched, extracellular lamellae, but also in corneodesmosome structures within the SC interstices. This problem has been overcome by the development and widespread deployment of ruthenium tetroxide (RuO4) postfixation, which resolves key ultrastructural features of the SC extracellular matrix. The failure to include RuO4-postfixed material in the evaluation of the DOC would be analogous to attempting to diagnose the blistering diseases without the ability to view components of the epidermal basement membrane.
Fig. 1. The SC. a ‘Normal basket weave’ = artifact of lipid extraction during tissue processing. b Frozen section stained with hydrophobic dye demonstrating that membrane domains in the SC are neutral and lipid enriched. SG = Stratum granulosum.
In subsequent sections, we will review the subcellular consequences of many of the genetically characterized DOC, utilizing ultrastructural features captured by the application of a battery of techniques, including (but not limited to) RuO4 postfixation. In many cases, we show further the impact of these changes for permeability barrier homeostasis. These efforts are still a work in progress, not only because some of the disorders have not yet been characterized at a molecular level, but also because many have not been evaluated using current morphological methods. Nevertheless, many as yet unpublished, potentially diagnostic observations are presented for the first time in this volume, which shed further light on the pathogenesis of several DOC. These ultrastructural studies include Refsum disease, CHILD (congenital hemidysplasia with ichthyosiform erythroderma and limb defects) syndrome, Sjögren-Larsson syndrome, ichthyosis vulgaris, IPS and ichthyosis en confettis.
In Appendix 1, we provide protocols for proper tissue handling, primary fixation, postfixation (OsO4 and RuO4), cytochemical and tracer methods, with the intent to spur future efforts to explore the pathogenesis of this fascinating but complex group of disorders. Finally, and most importantly, we believe that this effort is not merely a ‘stamp collection’ - in understanding how the epidermis fails in these genetic diseases, one can shed new light not only on disease pathogenesis, but also on norma...