![]()
III. The Quest for Achille’s Heel of Leishmania. Singular Targets as New Avenues for Drug Development
![]()
CHAPTER 12
Addressing the Molecular Biology of Leishmania for Drug Development
BRIANNA NORRIS-MULLINS AND MIGUEL A. MORALES*
Eck Institute for Global Health, Department of Biological Sciences, University of Notre Dame, Notre Dame, IN 46556, USA
12.1 Introduction
Kinetoplastid parasites of the genus Leishmania are the causative agents of leishmaniasis, a vector-borne disease that affects millions of people worldwide.1–3 Globally, approximately 310 million people are at risk of acquiring the disease, which is endemic in over 90 countries spanning four continents.4 In its digenetic life cycle Leishmania parasites cycle between the sand fly vector, where they persist as promastigotes, and phagolysosomes of mammalian macrophages, where they survive as amastigotes. The ability of the parasites to survive these distinct environments is dependent on the developmental regulation of numerous genes, whose identification may result in therapeutic targets.5 Though there is currently no human safe vaccine for leishmaniasis, some treatments are available and are, typically, species-specific. Current existing therapies for leishmaniasis are inadequate due to resistance, safety and cost, underscoring the necessity for safer therapies with alternate modes of action. Leishmaniasis affects the poorest sectors of low-income countries, therefore, public health therapies to combat the disease have little prospect of major economic return for private industry. Consequently, there are few large-scale drug discovery programs to identify new chemical entities to combat leishmaniasis.4,6,7 In implementing effective control strategies against leishmaniasis, it is vital to have a thorough understanding of the host–pathogen–vector relationship and, more importantly, of their interactions at a molecular level.8,9 Recent advances in ‘omics’ technologies, including genomics, transcriptomics and proteomics, make available the tools necessary for advancing our current understanding of the molecular biology of Leishmania9 with the purpose of exploiting this information for the development of new alternatives for anti-leishmanial therapeutics.
12.2 The Leishmania Genome
In 2005, the first ever sequenced genomes of Leishmania major10 and two distantly related kinetoplastid protozoa, Trypanosoma brucei11 and Trypanosoma cruzi,12 revealed large-scale genetic preservation.13 Despite a conserved core of genes, more than 1000 Leishmania-specific genes were identified; many of which remain uncharacterized.14 In 2007, genomic studies were extended to two additional Leishmania species: Leishmania infantum and Leishmania braziliensis. Upon bioinformatics analysis, more than 99% gene conservation between the three Leishmania genomes (L. major, L. infantum and L. braziliensis) was identified, yielding roughly 200 differences at the gene or pseudogene level with only a fraction of these being species-specific.14 The genomes of Leishmania amazonensis, Leishmania mexicana, Leishmania donovani and Leishmania tarentolae have since been sequenced, and comparative analyses have shown similar results.9,15 While findings indicate that minimal species-specific genes are important for pathogenesis, the underlying profound and unexpected conclusion is that the parasite genome plays a very small part in determining disease clinical presentation;14 this makes it nearly impossible to use species-specific genes for the development of therapeutic agents aimed at treating distinct clinical manifestations caused by Leishmania spp.
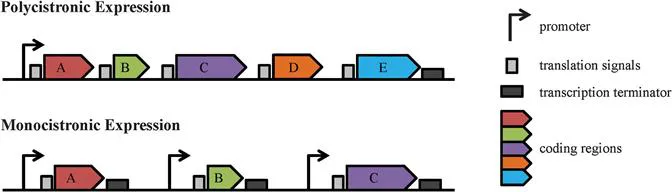
Leishmania parasites exhibit unique biological features ranging from gene organization, through transcription and mRNA processing, to post-translational modifications.5,16 For example, at the genomic level, many Leishmania genes are arranged into long, strand-specific, polycistronic clusters (Figure 12.1).10,17,18 This major difference between Leishmania spp. and higher eukaryotes has been extensively studied, has potential therapeutic implications for drug discovery and will be discussed in detail in the following section.
Figure 12.1 Gene Expression in Trypanosomatids vs. Higher Eukaryotes. In trypanosomatid parasites, large clusters of unrelated genes (coding regions) are organized as polycistronic transcription units (PTUs) under the control of a single promoter and termination codon (top). This differs from higher eukaryotes, where each individual gene has its own set of transcription signals (bottom).
12.3 The Leishmania Transcriptome
Although comparing the sequenced genomes of Leishmania species provides a solid basis for beginning to understand mechanisms of host–parasite interactions, it seems likely that a better understanding of disease pathogenesis could be gained by studying the regulation of gene expression during different parasite life cycle stages.9 The advent of high-throughput transcriptomic technologies, such as RNA sequencing, have facilitated such studies, however, the peculiar mechanisms of Leishmania gene expression partially dampen the enthusiasm for large-scale transcriptomic analyses. Indeed, most Leishmania genes have been shown to be constitutively expressed throughout the life cycle. It is important to note, though, that the noticeable variation in chromosome and gene copy numbers among L. infantum, L. braziliensis and L. major indicates that genome plasticity, rather than differential expression of single genes, could be the key to the different tissue tropism of Leishmania spp.9
12.3.1 Polycistronic Transcription and Trans-splicing Mechanisms
Similar to other eukaryotic organisms, Leishmania parasites use three different types of RNA polymerases (pol) to transcribe their genes. RNA pol I and RNA pol III are responsible for transcription of ribosomal RNA (rRNA) and transfer RNA (tRNA) genes, respectively, while RNA pol II transcribes protein-coding genes.19 It has been suggested, however, that in Leishmania, RNA pol II does not regulate transcription; this is due to the lack of transcription initiation sites for protein-coding genes within the organism.20 Furthermore, common transcription facto...