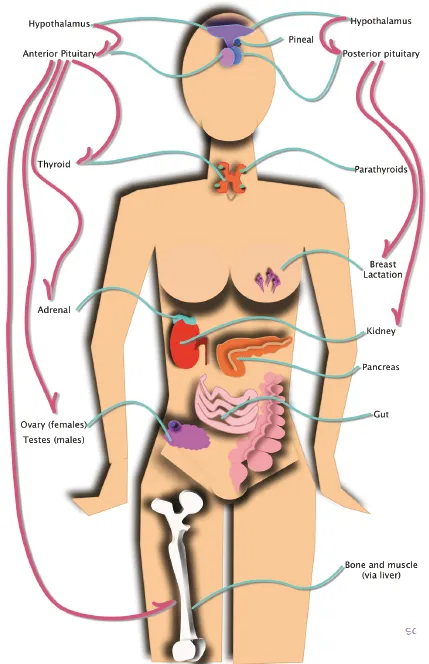
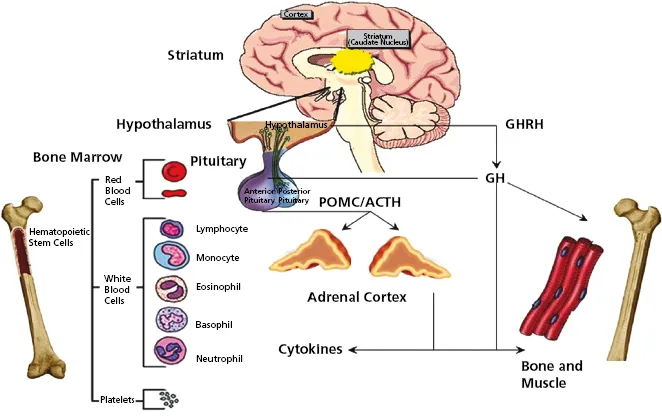
Do you want to be up to date on the latest concepts of diagnosis and treatment of patients suffering from disorders of the pituitary gland?
Are you looking for an expert guide to the best clinical management?
If so, this is the book for you, providing a full analysis of pituitary disorder management from acromegaly to Addison's Disease; from Cushing's Disease to hypopituitarism; from hormone disorders to hormone replacement.
Well-illustrated throughout, and with contributions from leading specialists in pituitary disease, inside you'll find comprehensive and expert coverage, including:
- Diagnosing pituitary disease
- Management options for each disorder
- Complications that can occur
- Psychological and psychosocial effects of pituitary disease
- What outcomes you and your patients can expect over the long term
- Current research and clinical trials related to pituitary disease
Pituitary Disorders: Diagnosis and Management is the perfect clinical tool for physicians and health care providers from many related disciplines, and an essential companion for the best quality management of pituitary patients.