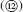![]()
Chapter 1
Introduction
“The eternal mystery of the world is its comprehensibility. The fact that it is comprehensible is a miracle.”
—Albert Einstein
The aim of this chapter is to discuss the whole picture of oral absorption of a drug in a comprehensive and descriptive manner without using any mathematical equation.
1.1 An Illustrative Description of Oral Drug Absorption: The Whole Story
The oral absorption of a drug is a sequential process of dissolution and intestinal membrane permeation of a drug in the gastrointestinal (GI) tract (Fig. 1.1).
After dosing a drug product (e.g., tablet and capsule), the formulation disintegrates to release solid particles of active pharmaceutical ingredient (API)
in
Fig. 1.1). The released API particles then dissolve into the GI fluid as molecularly dispersed drug molecules
. The maximum amount of a drug dissolved in the GI fluids is limited by the solubility of the drug in the fluids. In some cases, after an initial API form (such as a salt form) being dissolved, a transient supersaturated state is produced, and then, another solid form (i.e., a free base or an acid) can precipitate out in the intestinal fluid via nucleation
. The dissolved drug molecules are conveyed close to the intestinal wall by the macromixing of the intestinal fluid
and further diffuse through the unstirred water layer (UWL), which is adjacent to the epithelial cellular membrane
. The drug molecules then permeate the apical membrane of the epithelial cells mainly by passive diffusion
but in some cases, via a carrier protein (a transporter) such as PEP-T1
. If the drug is a substrate for an efflux transporter such as P-gp, a portion of the drug molecules is carried back to the apical side
. Some drugs pass through the intercellular junction (the paracellular route)
. In the epithelial cells, the drug could be metabolized by enzymes such as CYP3A4
. After passing through the basolateral membrane
, the drug molecules reach the portal vein. The drug molecules in the portal vein then pass through the liver and reach the systemic circulation
.
1.2 Three Regimes of Oral Drug Absorption
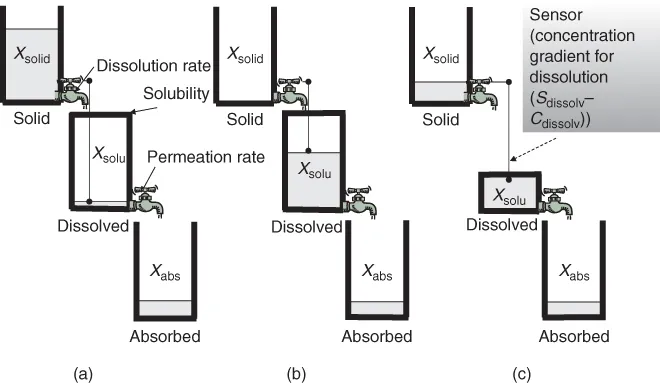
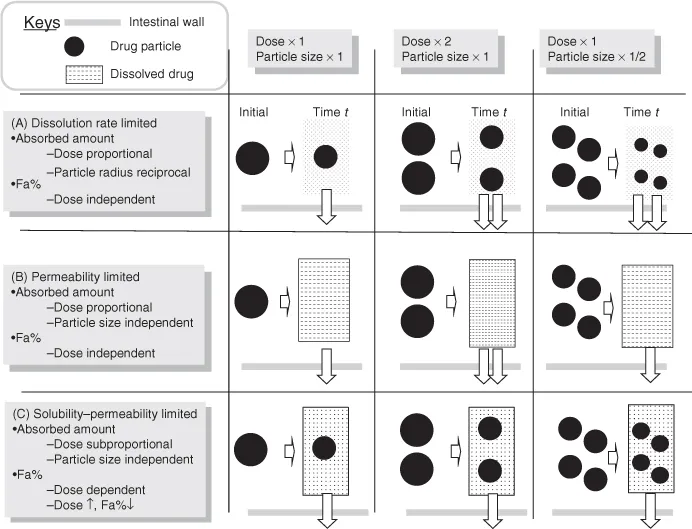
The central dogma of oral drug absorption is the interplay between solubility, the dissolution rate and permeability of a drug. On the basis of the central dogma, the three rate-limiting steps of oral absorption can be defined. Crystal clear understanding of these regimes is the first step toward understanding biopharmaceutical modeling [1]. Figure 1.2 shows the schematic presentation of the rate-limiting steps in the oral absorption of a drug [2].
- Dissolution rate-limited absorption (DRL) (Fig. 1.2a)
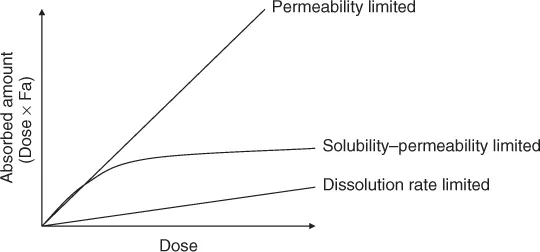
— In this case, the dissolution rate of API is much slower than the permeation rate. Once the API is dissolved, the drug molecules instantly permeate the intestinal membrane and get absorbed into the body. The dissolved drug molecules does not accumulate in the intestinal fluid as it is rapidly removed by the intestinal membrane permeation. Therefore, the dissolved drug concentration (Cdissolv) in the intestinal fluid is maintained well below the saturated solubility of a drug (Sdissolv). In this case, the rate of drug absorption is determined by the dissolution rate. The fraction of a dose absorbed (Fa%) is not dependent on the dose strengths of a drug (Figs. 1.2a, 2.3), whereas particle size reduction will be effective in increasing Fa% (Fig. 1.4).
- Permeability-limited absorption (PL) (Fig. 1.2b)
— In this case, the API dissolves immediately and completely in the intestinal fluid; however, the permeation of the drug is slow. Owing to the slow permeation clearance, the dissolved drug molecules accumulate in the intestinal fluid. The dissolved drug concentration does not reach its saturated solubility when the administered drug amount (Dose) is smaller than the solubilization capacity of the intestinal fluid (Dose < Sdissolv × VGI (intestinal fluid volume)) (Fig. 1.2b). In this case, the rate of drug absorption is determined by the permeation rate. Fa% is not dependent on the dose strength and particle size of a drug (Fig. 1.4).
- Solubility-permeability-limited absorption (SL) (Fig. 1.2c)
— When the dissolution rate of a drug is much faster than the permeation rate and the solubilization capacity of the intestinal fluid is smaller than the dose strength (Dose > Sdissolv × VGI), the dissolved drug molecules accumulate in the intestinal fluid and the dissolved drug concentration reaches the saturated solubility of the drug. In this case, the total absorption flux is determined as the maximum amount of dissolved drug (= Sdissolv × VGI) multiplied by the permeation rate of the drug (Fig. 1.2c). This case is further categorized by the rate-limiting step in the permeation process, that is, solubility–epithelial membrane permeability limited (SL-E) and solubility-UWL permeability limited (SL-U) cases [3]. Fa% decreases as the dose strength increases (Fig. 1.4).1 Particle size reduction will not be effective in increasing Fa% for SL-E cases but could be effective for SL-U cases (Section 4.7.2).
The balance of the dissolution rate coefficient (kdiss), the permeation rate coefficient (kperm) and the ratio of dose strength to the solubilization capacity of the GI fluid (Dose/Sdissolv × VGI) determines the regime of oral drug absorption. The last parameter is called the dose number (Do). The dose number is one of the most important parameters in biopharmaceutical modeling.
1.3 Physiology of the Stomach, Small Intestine, and Colon
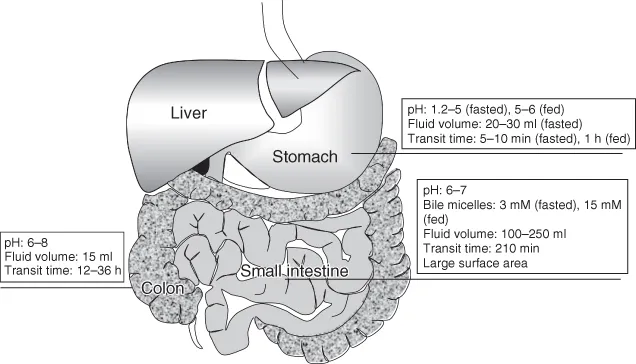
The GI tract can be roughly divided into the stomach, the small intestine, and the colon (Fig. 1.5). In humans, the pH of the stomach fluid is 1.2–2.5 in the fasted state but 5–6 in the fed state. The fluid volume in the stomach is ca. 30 ml. The pH of the intestinal fluid is 6.0–7.0 and is maintained relatively constant. The fluid volume in the small intestine is ca. 100–250 ml. Bile acid concentration in the small intestine is ca. 3 mM in the fasted state and 10–15 mM in the fed state. The pH of the colonic fluid is 6.0–8.0. The fluid volume in the colon is ca. 15 ml.
Drug absorption mainly occurs in the small intestine as it has the largest absorptive surface area and the largest fluid volume in the GI tract. Bile micelles can enhance the solubility and dissolution rate of a lipophilic drug. The stomach pH can affect the solubility and dissolution of a free base and its salt. It can also affect the precipitation of free acid from its salt. For low permeability and/or low solubility drugs, colonic absorption is usually negligible because of the small absorptive surface area, small fluid volume, solidification of the fluid, lack of bile micelles, etc.
1.4 Drug and API Form
The patterns of oral drug absorption can be also categorized from the viewpoint of the proper...