![]()
CHAPTER 1
Introduction to Randomized Clinical Trials in Cardiovascular Disease
Tobias Geisler,1 Marcus D. Flather,2 Deepak L. Bhatt,3 and Ralph B. D’Agostino, Sr4
1University Hospital Tübingen, Tübingen Medical School, Tübingen, Germany
2University of East Anglia and Norfolk and Norwich University Hospital, Norwich, UK
3VA Boston Healthcare System; Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, USA
4Boston University, Boston, MA, USA
What is a Randomized Clinical Trial?
The question “does it work” is common when a treatment is being considered for a patient. How do we know whether treatments “work” and what is the best way to demonstrate the efficacy and safety of new treatments? The main rationale behind a clinical trial is to perform a prospective evaluation of a new treatment in a rigorous and unbiased manner to provide reliable evidence of safety and efficacy. This is done by comparing the new treatment to a comparator or control treatment. Defining the term “clinical trial” is not as straightforward as it seems. In its simplest form, a clinical trial is any comparative evaluation of treatments involving human beings. Randomized clinical trials (RCTs) are the optimal means we use to achieve this demonstration. In this chapter we explore the relevance of RCTs to modern medicine and review strengths and weaknesses of this methodology (Table 1.1). As we will discuss below, RCTs represent the highest form of a clinical trial. Since the results of RCTs inform clinical practice guidelines, it is increasingly important for clinicians to understand their methodology, including their strengths and weaknesses. In this chapter we provide an overview of the main methodological aspects of well-designed RCTs.
Table 1.1 Issues for design/conduct and analysis of randomized clinical trials.
- Study objective
- Study populations
- Efficacy variables
- Control groups
- Study design (bias)
- Study design (samples)
- Comparisons
- Trial monitoring
- Data analysis sets
| - Unit of analysis
- Missing data
- Analysis methods
- Sample size/power
- Safety
- Subsets and more
- Number of studies
- Clinical significance
|
Concept of Randomization
The RCT is the most powerful design to prove whether or not there is a valid effect of a therapeutic intervention compared to a control. Randomization is a process of allocating treatments to groups of subjects using the play of chance. It is the mechanism that controls for factors except for the treatments, and allows comparison of the treatment under investigation with the control in an unbiased manner. It is important that information on the process of randomization is included in the trial protocol. The number of subjects allocated to each group, those who actually received the assigned treatment and reasons for non-compliance need to be recorded. In a representative analysis of trials listed in the free MEDLINE reference and abstract database at the United States National Library of Medicine (PubMed) in 2000, an adequate approach to random sequence generation was reported in only 21% of the trials [1]. This increased to 34% for a comparable cohort of PubMed-indexed trials in 2006 [2].
The procedure to assign interventions to trial participants is a critical aspect of clinical trial design. Randomization balances for known and unknown prognostic factors (covariates) allows the use of probability theory to express the likelihood that any difference in outcome between intervention groups merely reflects chance [3]. It facilitates blinding the identity of treatments to the investigators, participants, and evaluators, possibly by use of a placebo, which reduces bias after assignment of treatments [4]. Successful randomization is dependent on two related elements—generation of an unpredictable allocation sequence and concealment of that sequence until assignment takes place [5].
There are many procedures for randomization in the setting of a clinical trial and these will be discussed in detail below [see Study design (bias)]. For now we call attention to its importance in allowing the unbiased comparison of the investigational treatment and a control in a clinical trial.
Clinical Trial Phases
Preclinical Studies
Preclinical studies of potentially useful treatments are usually carried out to understand mechanisms of action, effect of different doses, and possible unwanted effects. There are two main types of preclinical studies—those using whole animal models and those using components of living tissue, usually cells or organs. Preclinical studies help to build up hypotheses about how and why treatments may work. Most of these experiments are not randomized and there may be substantial reporting bias (i.e., only interesting results are reported), but they are an essential step in the development of new treatments.
Phase 1 Clinical Trials
The first step to evaluate the safety of a new drug or biological substance after successful experiments in animals is to evaluate how well it can be tolerated in a small number of individuals. This phase is intended to test the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of a drug. Although it does not strictly meet the definition criteria of a clinical trial, this phase is often termed a phase 1 clinical trial. Usually, if the drug has a tolerable toxicological profile, a small number of healthy volunteers are recruited. If the drug has an increased toxicological profile, often critically ill patients are included in whom standard, guideline-based therapy fails. The design of phase 1 clinical trial is usually simple. In general, drugs are tested at different doses to determine the maximum tolerated dose (MTD) before signs of toxicity occur. The most difficult challenge in the planning of phase 1 trials is finding ways to adequately translate the animal experimental data into a dosing scheme and not to exceed the maximum tolerated dose in humans. Phase 1 clinical trials are dose-ranging studies to identify a tolerable dose range that can be evaluated further for safety in phase 2 trials. There are different ways to adjust doses in a phase 1 clinical trial, e.g., single ascending and multiple ascending dosing schemes. Studies in apparently healthy human volunteers usually involve short exposure to new treatments to understand the effects of different doses on human physiology. Starting at low or subtherapeutic doses, especially with novel immunogenic agents, is essential to ensure that unexpected serious side effects are reduced.
Phase 2 Clinical Trials
Phase 2 clinical trials refer to the results of phase 1 trials. Once the maximum tolerated dose has been defined and an effective and tolerable dose range has been determined, phase 2 trials are designed to investigate how well a drug works in a larger set of patients (usually 100–600 subjects and sometimes up to 4000 patients, depending on the number of groups to be investigated) and to continue measurements of PK and PD in a more global population. Some phase 2 trials are designed as case series where selected patients all receive the drug or as randomized trials where candidate doses of a drug are tested against placebo. Usually, different doses of a pharmacological treatment will be compared against placebo in a randomized study design with outcomes based on the mechanistic action of the treatment being evaluated. For example, phase 2 trials of anticoagulants will usually document laboratory measures of anticoagulant effect, incidence of major and minor bleeding, and effects on relevant clinical outcomes. Minimizing risk to patients is essential as most treatments evaluated in phase 2 trials will never be approved for human use. Strategy-based treatments such as new methods for percutaneous coronary intervention (PCI) or surgical procedures also have their equivalent “phase 2” trials in which the new techniques are systematically tested in smaller number of patients to ensure safety and feasibility before being tested in larger trials. For obvious reasons these trials cannot be “placebo controlled,” but should compare the new strategy with an established one. Sometimes “phase 2” trials of treatment strategies are not randomized, which often makes it difficult to draw conclusions about safety and feasibility, and to plan further larger trials.
As an example, in the phase 2 trial Anti-Xa Therapy to Lower cardiovascular events in Addition to standard therapy in Subjects with Acute Coronary Syndrome–Thrombolysis in Myocardial Infarction 46 (ATLAS-1-TIMI 46 trial), the oral factor Xa inhibitor rivaroxaban was tested in several doses (5 mg, 10 mg, or 20 mg total daily dose, given either once or twice daily) in a total of 3491 patients with acute coronary syndromes (ACS) being treated with aspirin or aspirin and clopidogrel and compared with placebo. There was a dose-related increase in bleeding and a trend toward a reduction in ischemic events with the addition of rivaroxaban to antiplatelet therapy in patients with recent ACS. The researchers found that patients assigned to 2.5 mg and 5.0 mg twice-daily rivaroxaban in both the aspirin alone and aspirin plus clopidogrel groups had the most efficacious results versus placebo [6]. These results led to a selection of these dosing groups for transition into a large phase 3 trial that enrolled 15 526 patients (ATLAS-2-TIMI-51) [7].
Phase 3 Clinical Trials
Phase 3 trials are usually RCTs, often multicenter, and including up to several thousand patients (the sample size depending upon the disease and medical condition being investigated). Due to the study size and duration, phase 3 trials are the most expensive, time-consuming, and complex trials to design and run, especially in therapies for chronic medical conditions, and are usually the “pivotal” trials for registration and marketing approval. Other possible motives for conducting phase 3 trials include plans to extend the label by the sponsor (i.e., to demonstrate the drug is effective for subgroups of patients/disease conditions beyond the use for which the drug was originally approved); to collect additional safety data; or to secure marketing claims for the drug. Trials at this stage are sometimes classified as “phase 3B trials” in contrast to “phase 3A trials,” denoting RCTs performed before marketing approval [8]. Once a drug has proved acceptable in phase 3 trials, the trial results are usually combined into a large comprehensive document describing the methods and results of animal (preclinical) and human (clinical studies), manufacturing processes, product characteristics (e.g., formulation, shelf-life). This document serves as a “regulatory submission” to be reviewed by the appropriate regulatory authorities in different countries before providing approval to market the drug.
Phase 4 Clinical Trials
In phase 4 trials, post-marketing studies delineate additional information, including the drug’s risks, benefits, and optimal use. They also aim to see if a treatment or medication can be used in other circumstances beyond the originally approval indications. Phase 4 clinical trials are done after a treatment has gone through all the other phases and is already approved by the regulatory health authorities. Phase 4 clinical trials may not necessarily be RCTs. A large body of phase 4 trials is made up of registries and observational studies.
The following discussion about the methodology will mainly focus on phase 3 confirmatory RCTs.
Study Objective
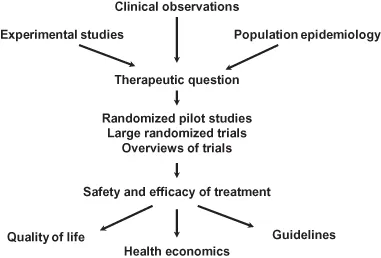
The search for new treatments is an evolutionary process, starting with a series of questions and eventually providing answers through a complex route that involves epidemiology (pattern and impact of disease in the population), basic science (cellular, mechanical, and genetic nature of the disease), and clinical trials to understand the response of patients to the new treatment. Trials that show clear benefits of treatments are usually followed by an assessment of cost and “affordability” to understand if the new treatment can actually be used in clinical practice. Some of these pathways are illustrated in Figure 1.1.
The quest to find effective and safe treatments arises from the needs of patients who present with illness and suffering. Thus, most clinical research is responsive in nature; we are not trying to improve on the healthy human but rather to treat and prevent illness and disease. However, in order to find an effective treatment, it is essential to understand the cause and pathology of the disease. Once specific causes are identified, whether they are protein deficiencies, transport errors, metabolic problems or genetic defects, it becomes possible to identify potential treatments that can then be tested in clinical trials. The challenge is that clinical trials take time and are costly to run, which means that they should be reserved for clinically important questions. Most clinical trials are set up and run by industry for commercial gain—often as industry/academic partnerships—but it should be emphasized that important health issues should be supported by the major healthcare providers, including governments and insurance agencies as part of their programs to improve health [1]. At present, most independent, non-commercial medical research is funded by competitive grants from governments or charities. While the competitive process helps to maintain high standards, it is an unpredictable method of funding and can lead to delays in carrying out important clinical trials. Lastly, well-intentioned but bureaucratic regulations applied to medical research are actually leading to substantial delays in important and effective treatments reaching patients in a timely manner. Thus, randomized trials are needed as the final pathway to test the hypothesis “Does it work?”. To answer this question reliably, large trials involving many patients from many centers are needed, which means that trial procedures including data collection and analysis need to be as simple and streamlined as possible [9,10].
Given all the above, when a specific phase 3 clinical trial is being designed, the first question is “What is ...