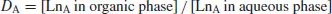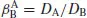![]()
1
Introduction to the Lanthanides
By the end of this chapter you should be able to:
- understand that lanthanides differ in their properties from the s- and d-block metals;
- recall characteristic properties of these elements;
- appreciate reasons for their positioning in the Periodic Table;
- understand how the size of the lanthanide ions affects certain properties and how this can be used in the extraction and separation of the elements;
- understand how to obtain pure samples of individual Ln3+ ions.
1.1 Introduction
Lanthanide chemistry started in Scandinavia. In 1794 Johann Gadolin succeeded in obtaining an ‘earth’(oxide) from a black mineral subsequently known as gadolinite; he called the earth yttria. Soon afterwards, M.H. Klaproth, J.J. Berzelius and W. Hisinger obtained ceria, another earth, from cerite. However, it was not until 1839–1843 that the Swede C.G. Mosander first separated these earths into their component oxides; thus ceria was resolved into the oxides of cerium and lanthanum and a mixed oxide ‘didymia’ (a mixture of the oxides of the metals from Pr through Gd). The original yttria was similarly separated into substances called erbia, terbia, and yttria (though some 40 years later, the first two names were to be reversed!). This kind of confusion was made worse by the fact that the newly discovered means of spectroscopic analysis permitted misidentifications, so that around 70 ‘new’ elements were erroneously claimed in the course of the century.
Nor was Mendeleev’s revolutionary Periodic Table a help. When he first published his Periodic Table in 1869, he was able to include only lanthanum, cerium, didymium (now known to have been a mixture of Pr and Nd), another mixture in the form of erbia, and yttrium; unreliable information about atomic mass made correct positioning of these elements in the table difficult. Some had not yet been isolated as elements. There was no way of predicting how many of these elements there would be until Henry Moseley (1887–1915) analysed the X-ray spectra of elements and gave meaning to the concept of atomic number. He showed that there were 15 elements from lanthanum to lutetium (which had only been identified in 1907). The discovery of radioactive promethium had to wait until after World War 2.
It was the pronounced similarity of the lanthanides to each other, especially each to its neighbours (a consequence of their general adoption of the +3 oxidation state in aqueous solution), that caused their classification and eventual separation to be an extremely difficult undertaking.
Subsequently it was not until the work of Bohr and of Moseley that it was known precisely how many of these elements there were. Most current versions of the Periodic Table place lanthanum under scandium and yttrium.
1.2 Characteristics of the Lanthanides
The lanthanides exhibit a number of features in their chemistry that differentiate them from the d-block metals. The reactivity of the elements is greater than that of the transition metals, akin to the Group II metals:
1. A very wide range of coordination numbers (generally 6–12, but numbers of 2, 3 or 4 are known).
2. Coordination geometries are determined by ligand steric factors rather than crystal field effects.
3. They form labile ‘ionic’ complexes that undergo facile exchange of ligand.
4. The 4f orbitals in the Ln3+ ion do not participate directly in bonding, being well shielded by the 5s2 and 5p6 orbitals. Their spectroscopic and magnetic properties are thus largely uninfluenced by the ligand.
5. Small crystal-field splittings and very sharp electronic spectra in comparison with the d-block metals.
6. They prefer anionic ligands with donor atoms of rather high electronegativity (e.g. O, F).
7. They readily form hydrated complexes (on account of the high hydration energy of the small Ln3+ ion) and this can cause uncertainty in assigning coordination numbers.
8. Insoluble hydroxides precipitate at neutral pH unless complexing agents are present.
9. The chemistry is largely that of one (3+) oxidation state (certainly in aqueous solution).
10. They do not form Ln=O or Ln≡N multiple bonds of the type known for many transition metals and certain actinides.
11. Unlike the transition metals, they do not form stable carbonyls and have (virtually) no chemistry in the 0 oxidation state.
1.3 The Occurrence and Abundance of the Lanthanides
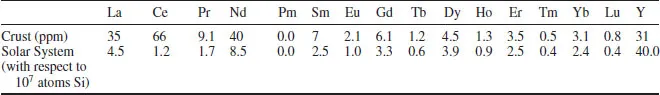
Table 1.1 presents the abundance of the lanthanides in the earth’s crust and in the solar system as a whole. (Although not in the same units, the values in each list are internally consistent.)
Two patterns emerge from these data. First, that the lighter lanthanides are more abundant than the heavier ones; secondly, that the elements with even atomic number are more abundant than those with odd atomic number. Overall, cerium, the most abundant lanthanide on earth, has a similar crustal concentration to the lighter Ni and Cu, whilst even Tm and Lu, the rarest lanthanides, are more abundant than Bi, Ag or the platinum metals.
Table 1.1 Abundance of the lanthanides
The abundances are a consequence of how the elements were synthesized by atomic fusion in the cores of stars with heavy elements only made in supernovae. Synthesis of heavier nuclei requires higher temperature and pressures and so gets progressively harder as the atomic number increases. The odd/even alternation (often referred to as the Oddo–Harkins rule) is again general, and reflects the facts that elements with odd mass numbers have larger nuclear capture cross sections and are more likely to take up another neutron, so elements with odd atomic number (and hence odd mass number) are less common than those with even mass number. Even-atomic-number nuclei are more stable when formed.
1.4 Lanthanide Ores
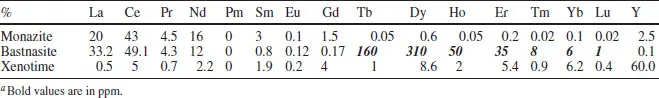
Principal sources (Table 1.2) are the following:
Bastnasite LnFCO3; Monazite (Ln, Th)PO4 (richer in earlier lanthanides); Xenotime (Y, Ln)PO4 (richer in later lanthanides). In addition to these, there are Chinese rare earth reserves which amount to over 70% of the known world total, mainly in the form of the ionic ores from southern provinces. These Chinese ion-absorption ores, weathered granites with lanthanides adsorbed onto the surface of aluminium silicates, are in some cases low in cerium and rich in the heavier lanthanides (Longnan) whilst the Xunwu deposits are rich in the lighter metals; the small particle size makes them easy to mine. The Chinese ores have made them a leading player in lanthanide chemistry.
Table 1.2 Typical abundance of the lanthanides in oresa
1.5 Extracting and Separating the Lanthanides
These two processes are not necessarily coterminous. Whilst electronic, optical and magnetic applications require individual pure lanthanides, the greatest quantity of lanthanides is used as mixtures, e.g. in mischmetal or oxide catalysts.
1.5.1 Extraction
After initial concentration by crushing, grinding and froth flotation, bastnasite is treated with 10% HCl to remove calcite, by which time the mixture contains around 70% lanthanide oxides. This is roasted to oxidize the cerium content to CeIV; on further extraction with HCl, the Ce remains as CeO2, whilst the lanthanides in the (+3) state dissolve as a solution of the chlorides.
Monazite is usually treated with NaOH at 150 °C to remove phosphate as Na3PO4, leaving a mixture of the hydrated oxides, which are dissolved in boiling HCl at pH 3.5, separating the lanthanides from insoluble ThO2. Sulfuric acid can also be used to dissolve the lanthanides.
1.5.2 Separating the Lanthanides
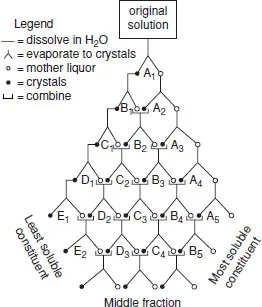
These can be divided into four types: chemical separations, fractional crystallization, ion-exchange methods and solvent extraction. Of these, only the last-named is used on a commercial scale (apart from initial separation of cerium). Chemical separations rely on using stabilities of unusual oxidation states; thus Eu2+ is the only ion in that oxidation state formed on reduction by zinc amalgam and can then be precipitated as EuSO4 (note the similarity with heavier Group 2 metals). Repeated (and tedious) fractional crystallization, which made use of slight solubility differences between the salts of neighbouring lanthanides, such as the bromates Ln(BrO3)3.9H2O, ethyl sulfates and double nitrates, were once the only possible way of obtaining pure lanthanides, as with the 15 000 recrystallizations carried out by the American C. James to get pure thulium bromate (1911) (Figure 1.1 indicates the principle of this method).
Figure 1.1 Diagrammatic representation of the system of fractional crystallization used to separate salts of the rare-earth elements (reproduced with permission from D.M. Yost, H. Russell and C.S. Garner, The Rare Earth Elements and their Compounds, John Wiley, 1947.)
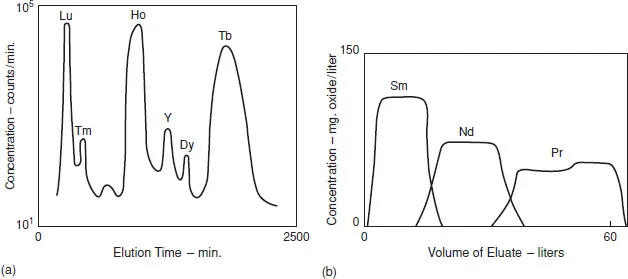
Ion-exchange chromatography is not of real commercial importance for large-scale production, but historically it was the method by which fast high-purity separation of the lanthanides first became feasible. As radioactive lanthanide isotopes are important fission products of the fission of 235U and therefore need to be separated from uranium, and because the actinides after plutonium tend to resemble the lanthanides, the development of the technique followed on the Manhattan project. It was found that if Ln3+ ions were adsorbed at the top of a cation-exchange resin, then treated with a complexing agent such as buffered citric acid, then the cations tended to be eluted in reverse atomic number order (Figure 1.2a); the anionic ligand binds most strongly to the heaviest (and smallest) cation, which has the highest charge density. A disadvantage of this approach when scaled up to high concentration is that the peaks tend to overlap (Figure 1.2b).
It was subsequently found that amine polycarboxylates such as EDTA4− gave stronger complexes and much better separations. In practice, some Cu2+ ions (‘retainer’) are added to prevent precipitation of either the free acid H4EDTA or the lanthanide complex HLn(EDTA).xH2O on the resin. The major disadvantage of this method is that it is a slow process for large-scale separations.
Figure 1.2 (a) Cation-exchange chromatography of lanthanides, (b) overlap of peaks at high concentration. (a) Tracer-scale elution with 5% citrate at pH 3.20 (redrawn from B.H. Ketelle and G.E. Boyd, J. Am. Chem. Soc., 1947, 69, 2800). (b) Macro-scale elution with 0.1% citrate at pH 5.30 (redrawn from F.H. Spedding, E.I. Fulmer, J.E. Powell, and T.A. Butler, J. Am. Chem. Soc., 1950, 72, 2354). Reprinted with permission of the American Chemical Society © 1978.
Solvent extraction has come to be used for the initial stage of the separation process, to give material with up to 99.9% purity. In 1949, it was found that Ce4+ could readily be separated from Ln3+ ions by extraction from a solution in nitric acid into tributyl phosphate [(BuO)3PO]. Subsequently the process was extended to separating the lanthanides, using a non-polar organic solvent such as kerosene and an extractant such as (BuO)3PO or bis (2-ethylhexyl)phosphinic acid [[C4H9CH(C2H5)]2P=O(OH)] to extract the lanthanides from aqueous nitrate solutions. The heavier lanthanides form complexes which are more soluble in the aqueous layer. After the two immiscible solvents have been agitated together and separated, the organic layer is treated with acid and the lanthanide extracted. The solvent is recycled and the aqueous layer put through further stages.
For a lanthanide LnA distributed between two phases, a distribution coefficent DA is defined:
For two lanthanides Ln
A and Ln
B in a mixture being separated, a separation factor
can be defined, where
β is very close to unity for two adjacent lanthanides in the Periodic Table (obviously, the larger β is, the better the separation).
In practice this process is run using an automated continuous counter-current circuit in which the organic solvent flows in the opposite direction to the aqueous layer containing the lanthanides. An equilibrium is set up between the lanthanide ions in the aqueous phase and the organic layer, with there tending to be a relative enhancement of the concentration of the heavier lanthanides in the organic layer. Because the separation between adjacent lanthanides in each exchange is relatively slight, over a thousand exchanges are used (see Figure 1.3). This method affords lanthanides of purity up to the 99.9% purity level and is thus well suited to large-scale separation, the products being suited to ordinary chemical use. However, for electronic or spectroscopic use (‘phosphor grade’) 99.999% puri...