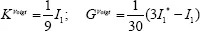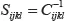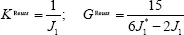![]()
Chapter 1
Bottom-Up: From Atoms to Concrete Structures 1
1.1. Introduction
More concrete is produced than any other synthetic material on Earth. The current worldwide cement production stands at 2.3 billion tons, enough to produce more than 20 billion tons or one cubic meter of concrete per capita per year. There is no other material that can replace concrete in the foreseeable future to meet the legitimate needs of our society’s housing, shelter, infrastructure, and so on. But concrete faces an uncertain future, due to a non-negligible ecological footprint that amounts to 5%–10% of the worldwide CO2 production. It now appears that mechanics can be the discipline that enables the development of a sustainable green concrete future.
We adopt here the perspective originating from Galileo’s Strength of Materials Theory that weight, and thus, CO2 emission, increases with the volume of the produced material, while strength of structural members increases with the cross-section. Hence, as one increases the strength of a material by a factor of x, the other reduces the environmental footprint by 1/x for pure compressive members such as columns and perfect arches and shells, x−2/3 for beams in bending and x−1/2 for slabs. Similarly, if one adopted a Linear Elastic Fracture model, an increase of the fracture toughness KIc = y KIc0 would entail a reduction of the environmental footprint by 1/y for columns and y−4/5 for (notched) beams in bending or in torsion. All this hints towards a critical role of mechanics, and in particular, strength of materials, fracture, and damage mechanics of concrete, in redesigning concrete materials and structures for the coming of age of global warming. In contrast to the classical top-down empirical approaches, we have chosen a bottom-up approach that starts at the electron and atomic scale to nanoengineer the fundamental building block of concrete, to assess the properties by nanoindentation, and to upscale strength, fracture, and stiffness properties from nanoscales to macroscales of day-to-day concrete engineering applications. The key to all this is the mechanics at the interface of physics and engineering. This chapter reviews some recent developments of this bottom-up approach.
1.2. A realistic molecular model for calcium-silicate-hydrates
The first step in setting up a bottom-up approach is to address the fundamental unit of concrete material behavior at electron and atomic scale. But, despite decades of studies of calcium-silicate-hydrate (C-S-H), the structurally complex binder phase of concrete and the interplay between chemical composition and density remain essentially unexplored. Together these characteristics of C-S-H define and modulate the physical and mechanical properties of this “liquid stone” gel phase.
1.2.1. Background
Much of our knowledge of C-S-H has been obtained from structural comparisons with crystalline C-S-H, based on HFW Taylor’s postulate that real C-S-H was a structurally imperfect layered hybrid of two natural mineral analogs: tobermorite of 14-Å interlayer spacing and jennite. While this suggestion is plausible in morphological terms, this model is incompatible with two basic characteristics of real C-S-H, specifically the calcium-to-silicon ratio (C/S) and the density. Recently, small-angle neutron scattering measurements have fixed the C/S ratio at 1.7 and the density at 2.6 g/cm3 [ALL 07], values that cannot be obtained clearly from either tobermorite (C/S = 0.83, 2.18 g/cm3) or jennite (C/S = 1.5, 2.27 g/cm3); see for instance [SHA 09]. From the standpoint of constructing a molecular model of C-S-H, this means that these crystalline minerals are not strict structural analogs. This brought about the development of a realistic molecular C-S-H, based on a bottom-up atomistic simulation approach that considers only the chemical specificity of the system as the overriding constraint [PEL 09]. By allowing for short silica chains distributed as monomers, dimers, and pentamers, this C-S-H archetype of a molecular description of interacting CaO, SiO2, and H2O units provides not only realistic values of the C/S ratio and the density computed by grand canonical Monte Carlo simulation of water adsorption at 300 K. The model, displayed in Figure 1.1, with a chemical composition of (CaO)1.65(SiO2)(H2O)1.75, also predicts other essential structural features and fundamental physical properties amenable to experimental validation. This model suggests that the C-S-H gel structure includes both glass-like short-range order and crystalline features of the mineral tobermorite.
1.2.2. Molecular properties of C-S-H
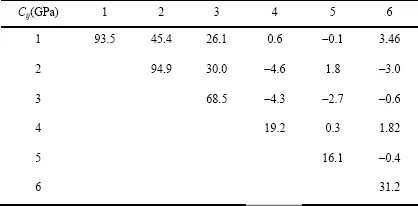
One of the great advantages of having a realistic molecular model of C-S-H is that it is possible to probe the structure mechanically. The first quantity of interest is the elasticity content of the molecular structure, which is given in Table 1.1 in the form of components of the elasticity tensor. As we may expect from a glassy-layered hybrid, the elasticity exhibits a high degree of anisotropy. For further applications, it will be useful to consider the random polycrystal properties, in the form of the Voigt—Reuss—Hill approximation classically used in mineralogy. As shown by Povolo and Bolmaro [POV 87], both the Voigt and Reuss models are built using the invariance of the trace of the 9 × 9 matrix representing the stiffness and compliance tensors, respectively. This leads to the observation (made by Hill) that the Voigt and Reuss averages only use 9 of the 21 independent elastic constants. Denoted by I1 = Ciijj and I1*=Cijij the traces (or linear invariants) of tensors Ciikl and Cijjl, respectively, the Voigt average is obtained from a comparison of those traces with their corresponding isotropic expressions, leading to:
Applying a similar procedure to the compliance tensor
, the Reuss average is obtained:
where J1 = Siijj and J1*=Sijij are the corresponding traces of the compliance tensors Siikl and Sijjl, respectively. The Reuss—Voigt—Hill polycrystal properties are obtained as the arithmetic mean of Reuss and Voigt bounds, which yields for C-S-H:
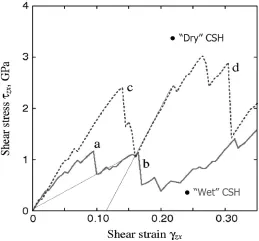
A second useful insight the molecular model can provide is about the strength behavior, by simulating for instance the stress-strain behavior of the C-S-H model in affine shear deformation (strain controlled) after first relaxing the computational cell using MD at 300 K, under constant NVT ensemble conditions. A series of shear strains in increments of 0.005 is imposed; after each increment the atomic configuration is relaxed and the shear stress determined from the virial expression. Figure 1.2 displays the shear stress-strain response of the C-S-H model. Two configurations are herein considered: the “wet” model, in which water is present particularly in the interlayer space, and the “dry” model, in which all water molecules have been removed. The stress-strain response shows that the presence of water in the interlayer space leads to a localization of deformation into a narrow band defined by a wet interlayer, akin to a fracturing or damage process. By contrast, the “dry” model shows a plastic deformation behavior, with irreversible (plastic) deformation upon unloading.
This shows that the...