![]()
Chapter 1
History of Drug Discovery
Jie Jack Li
1 Introduction
The history of drug discovery is as ancient as our glorious history of humanity. Shen Nung, The Divine Farmer, is fabled to have sampled 365 herbs himself to evaluate their medicinal value as early as 2337 B.C. in China. In 400 B.C. in Greece, Hippocrates, The Father of Medicine, decreed the Hippocratic Oath, in which a physician is to pledge “I will preserve the purity of my life and my arts”. Another Greek Physician, Galen (129–199), influenced 45 generations with his teachings of medicine, transforming medicine from art to science. During the Renaissance, Paracelsus (1493–1541) from Switzerland represented the pinnacle of Western medicine. Since then, a parade of luminaries began to unveil the myth of life. Andreas Vesalius (1514–1564) of Brussels founded the Science of Anatomy. William Harvey (1578–1657) of England made one of the greatest discoveries in medicine—Circulation of Blood. Dutchman Antonie von Leeuwenhoek (1632–1723) opened our eyes to a whole new world of microbes by inventing the microscope.
The intellectual contributions of these great men established the foundation of modern medicine and heralded the golden age of contemporary drug discovery.
2 Antibacterials
2.1 Lister and Carbolic Acid
Although many contributed to the germ theory, French chemist Louis Pasteur (1822-1895) transformed medicine with his well-designed experiments and a treatise entitled Organized Corpuscles Existing in Atmosphere published in 1855.1 Pasteur’s tour de force officially introduced and cemented germ theory within mainstream science. The pasteurization process, heating the food at a specific temperature for a certain time and then cooling it immediately, is still the standard practice. Merely one year later, Joseph Lister (1827–1912) in England successfully applied the germ theory by using carbolic acid (phenol) as an antiseptic during surgery to kill bacteria.
As a surgeon, Lister was appalled at the postsurgical infections, which killed patients with an astonishing 40–60% mortality rate. However, simple and direct application of the pasteurization process would not be acceptable during surgery—after all one could not simply boil the patient in hot water! Inspired by anecdotal success stories of carbolic acid (an ingredient isolated from coal tar, a waste from coal gas production) in deodorizing sewage and in controlling typhoid, Lister introduced carbolic acid as an antiseptic in surgery.2 It works by solubilizing the phospholipids in cell membranes, thus disrupting the cell membranes. Today, asepsis has largely replaced antisepsis in the operating rooms.
2.2 Dr. Ehrlich’s Magic Bullet
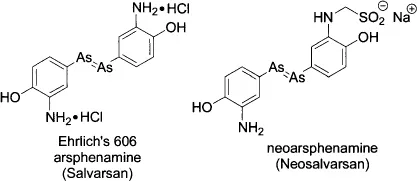
In the 1890s, together with Emil von Behring, Paul Ehrlich (1854–1915) developed a horse serum antitoxin to quell diphtheria. The vaccine saved thousands of children’s lives during the 1891 outbreak, for which he won the Nobel Prize in 1908. Since 1910, working with his Japanese associate Sachachio Hata, Ehrlich experimented with numerous chemicals to treat syphilis.3 They had some success with an arsenic compound atoxyl, which was efficacious but was too toxic. His chemist Alfred Bertheim (1879–1914) at first elucidated the chemical constitution of atoxyl and later synthesized innumerable arsenobenzene compounds including arsphenamine (Ehrlich’s 606), which was efficacious with an acceptable safety profile.4 Ehrlich licensed the drug to Hoeschst, who sold it under the trade name Salvarsan. To find less toxic and more water-soluble ant-syphilitics, Bertheim synthesized neoarsphenamine (Neosalvarsan). Both Salvarsan and Neosalvarsan had a tremendous impact on fighting syphilis, wiping out half of the syphilis infections in Europe in a mere five years (although syphilis was not completely eradicated until the introduction of penicillin in the 1940s).
Ehrlich was also the first to propose the side chain theory and the receptor theory to explain how drugs worked. While he is immortalized as the father of chemotherapy and with his concept of magic bullets, Bertheim, probably the first medicinal chemist in history, is largely forgotten. In addition to his important contributions to the discovery of Salvarsan and Neosalvarsan, Bertheim also published a book Ein Handbuch der organischen Arsenverbindungen (A Handbook of Organic Arsenic Compounds).
2.3 Domagk and Sulfa Drugs
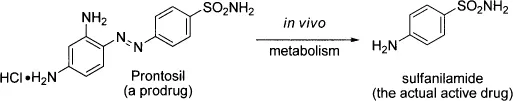
In 1932, Gerhard Domagk (1895–1964), the head of the Bacteriology Laboratory of I. G. Farbenindustrie Aktiengesellschaft (I. G. Farben), experimented with different dyes available to him in search of antibacterial drugs.5 Looking for antibacterials from dyes was most likely influenced by Ehrlich’s experiences with staining. By injecting dyes in mice infected with Streptococcus pyogenes bacterium, Domagk discovered that 2’,4’-diaminoazobenzene-4-sulfonamide, later branded as Prontosil, was effective in killing the bacterium without unacceptable toxic effects. The dye was prepared by Josef Klarer (1898–1953), a chemist at the Bayer Company, a branch of I. G. Farben. Later on, another Bayer chemist, Fritz Mietzsch (1896–1958), prepared the salt of Prontosil, enabling liquid formulation that was more amenable for injection. After Domagk’s first disclosure in 1935, Prontosil quickly became widely prescribed for streptococcal infections.
In 1935, a French husband and wife team, Professor Jacques Tréfouël and Madame Therèse Tréfouël, discovered that Prontosil was not active in vitro. The real active ingredient for its antibacterial activity is sulfanilamide (mistakenly being called sulfonamide even today), which is generated from in vivo metabolism of Prontosil. The mechanism of action (MOA) of sulfanilamides (antimetabolites) is through folate antagonism. Since the structure of sulfanilamide is similar to that of para-aminobenzoic acid (PABA), an essential ingredient for cell synthesis, it interrupts bacterial growth.
Domagk was bestowed the Nobel Prize in 1939. He received numerous letters from patients and doctors, expressing their gratitude for his discovery of Prontosil. In contrast, Klarer, the chemist who first synthesized it, received none.
2.4 Fleming, Florey, Chain, and Penicillin
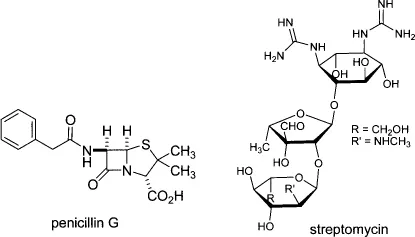
Alexander Fleming (1881–1955)6 actually discovered penicillin in 1928 in England, 4 years before Domagk’s Prontosil. However, more than 15 years elapsed until Howard Florey (1898–1968)7 and Ernst Chain (1907–1979)8 isolated enough penicillin and demonstrated its curative effects in both mice and humans. Penicillin quickly replaced Ehrlich’s 606 and Domagk’s sulfa drugs as the most widely used antibiotic. It works for Gram-positive bacterial infections, including strep and staph infections, pneumonia, gangrene, meningitis, as well as gonorrhea (now, however, a resistant form has emerged) and syphilis. The MOA of penicillin is through inhibition of cell wall synthesis. Because animals, including humans, lack a cell wall, penicillin exerts a bactericidal action selectively on growing or multiplying germs.
2.5 Waksman, Schatz, and Streptomycin
Inspired by Fleming’s success with penicillin, Selman A. Waksman (1888–1973), a professor of soil microbiology at Rutgers College, began to look for antibiotics in soil in 1939.9 At first his group isolated a small molecule antibiotic, actinomycin, and then streptothricin. Although both of them killed Gram-negative bacteria, they were so toxic that they also killed test animals. In October 1943, Waksman’s student Albert Schatz isolated streptomycin, an aminosugar. With assistance from Merck for large-scale production and the Mayo Clinic for animal testing and clinical trials, streptomycin was proven to be both safe and effective in treating tuberculosis. Astonishingly, only 3 years elapsed from its discovery to the first successful treatment of a human patient. Nowadays, it generally takes 12 years and over $1.3 billion to bring a drug to the market.
Streptomycin was the first drug to be effective against Gram-negative bacteria. It was particularly interesting at the time because of its activity against human tubercle bacillus, which made it the first specific agent effective in treating tuberculosis. Streptomycin works by inducing the binding of “wrong” tRNA-amino-acid complexes, resulting in synthesis of false protein.
2.6 Duggar, Conover, and Tetracyclines
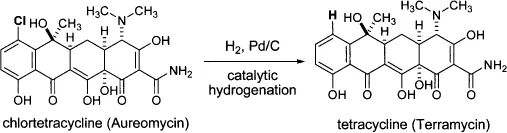
In 1945, 73 year old botanist Benjamin M. Duggar was a consultant for Lederle and led their screening efforts in the hunt for antibiotics. Coincidently, a sample from the University of Missouri, where Duggar taught botany 40 years earlier, yielded an antibiotic later named chlortetracycline. Lederle sold chlortetracycline under the brand name of Aureomycin in 1948. Nowadays, Benjamin Duggar is considered the pioneer of tetracycline antibiotics.10
In 1949, a yellow powder with strong antibiotic properties was isolated by Pfizer scientists from a soil sample. The soil organism was Streptomyces rimosus and the compound was generically known as oxytetracycline. Backtracking revealed that the soil sample was collected at the Terre Haute factory in Indiana owned by Pfizer, which later sold oxytetracycline under the brand name Terramycin.
Later on, Lloyd Conover at Pfizer stunned his colleagues by preparing another powerful antibiotic chemically from chlortetracycline. Under carefully controlled catalytic hydrogenation conditions, Conover converted Lederle’s chlortetracycline to tetracycline. That was the first example of a semisynthetic compound with antibiotic activities.
Tetracyclines are inhibitors of protein synthesis by inhibiting the binding of tRNA-amino-acid complexes. They are bacteriostatic.
2.7 Quinolones and Zyvox
In 1946, while pursuing better antimalarial drugs by synthesizing chloroquine, George Y. Lesher (1926–1990) at Sterling Winthrop Research Institute at Rensselaer isolated a by-product, nalidixic acid.11 It was found to be an antibacterial agent during routine screening. But it did not become popular until 1962 when Lesher introduced it into clinical practice for kidney infections. It was also used to treat urinary tract infections because it was excreted via urine in high concentration. Shortly after, the quinolone antibacterial field flourished, rendering thousands of 4-quinolone derivatives as represented by pipemidic acid. Nalidixic acid and pipemidic acid are considered the first-generation quinolone antibacterials. The drawbacks of these drugs were their moderate activity toward susceptible bacteria and poor absorption by the body.
In the early 1980s, fluorinated quinolone (fluoroquinolone) antibacterials were discovered to possess longer half-...