1.1 Enzymes Are Essential for Life
In high school biology classes life is often defined as “a series of chemical reactions.” This popular aphorism reflects the fact that living cells, and in turn multicellular organisms, depend on chemical transformations for every essential life process. Synthesis of biomacromolecules (proteins, nucleic acids, polysaccarides, and lipids), all aspects of intermediate metabolism, intercellular communication in, for example, the immune response, and catabolic processes involved in tissue remodeling, all involve sequential series of chemical reactions (i.e., biological pathways) to maintain life’s critical functions. The vast majority of these essential biochemical reactions, however, proceed at uncatalyzed rates that are too slow to sustain life. For example, pyrimidines nucleotides, together with purine nucleotides, make up the building blocks of all nucleic acids. The de novo biosynthesis of pyrimidines requires the formation of uridine monophosphate (UMP) via the decarboxylation of orotidine monophosphate (OMP). Measurements of the rate of OMP decarboxylation have estimated the half-life of this chemical reaction to be approximately 78 million years! Obviously a reaction this slow cannot sustain life on earth without some very significant rate enhancement. The enzyme OMP decarboxylase (EC 4.1.1.23) fulfills this life-critical function, enhancing the rate of OMP decarboxylation by some 1017-fold, so that the reaction half-life of the enzyme-catalyzed reaction (0.018 seconds) displays the rapidity necessary for living organisms (Radzicka and Wolfenden, 1995).
Enyzme catalysis is thus essential for all life. Hence the selective inhibition of critical enzymes of infectious organisms (i.e., viruses, bacteria, and multicellular parasites) is an attractive means of chemotherapeutic intervention for infectious diseases. This strategy is well represented in modern medicine, with a significant portion of antiviral, antibiotic, and antiparasitic drugs in clinical use today deriving their therapeutic efficacy through selective enzyme inhibition (see Table 1.1 for some examples).
TABLE 1.1 Selected Enzyme Inhibitors in Clinical Use or Trials
Source: Adapted and expanded from Copeland (2000).
| Acetazolamide | Carbonic anhydrase | Glaucoma |
| Acyclovir | Viral DNA polymerase | Herpes |
| Amprenavir, indinavir, nelfinavir, ritonavir, saquinavir | HIV protease | AIDS |
| Allopurinol | Xanthine oxidase | Gout |
| Argatroban | Thrombin | Heart disease |
| Aspirin | Cyclooxygenases | Inflammation, pain, fever |
| Amoxicillin | Penicillin binding proteins | Bacterial infection |
| Captopril, enalapril | Angiotensin converting enzyme | Hypertension |
| Carbidopa | Dopa decarboxylase | Parkinson’s disease |
| Celebrex, Vioxx | Cyclooxygenase-2 | Inflammation |
| CI-1040, PD0325901 | MAP kinase kinase | Cancer |
| Clavulanate | β-Lactamase | Bacterial resistance |
| Digoxin | Sodium, potassium ATPase | Heart disease |
| Efavirenz, nevirapine | HIV reverse transcriptase | AIDS |
| Epristeride, finasteride, dutasteride | Steroid 5α-reductase | Benign prostate hyperplasia, male pattern baldness |
| Fluorouracil | Thymidylate synthase | Cancer |
| Leflunomide | Dihydroorotate | Inflammation |
| Dehydrogenase | |
| Lovastatin and other statins | HMG-CoA reductase | Cholesterol lowering |
| Methotrexate | Dihydrofolate reductase | Cancer, immunosuppression |
| Nitecapone | Catechol-O-methyltransferase | Parkinson’s disease |
| Norfloxacin | DNA gyrase | Urinary tract infections |
| Omeprazole | H+, K+ ATPase | Peptic ulcers |
| PALA | Aspartate | Cancer |
| Transcarbamoylase | |
| Sorbinol | Aldose reductase | Diabetic retinopathy |
| Trimethoprim | Bacterial dihydrofolate reductase | Bacterial infections |
| Viagra, Levitra | Phosphodiesterase | Erectile dysfunction |
Although enzymes are essential for life, dysregulated enzyme activity can also lead to disease states. In some cases mutations in genes encoding enzymes can lead to abnormally high concentrations of the enzyme within a cell (overexpression). Alternatively, point mutations can lead to an enhancement of the specific activity (i.e., catalytic efficiency) of the enzyme because of structural changes in the catalytically critical amino acid residues. By either of these mechanisms, aberrant levels of the reaction product’s formation can result, leading to specific pathologies. Hence human enzymes are also commonly targeted for pharmacological intervention in many diseases.
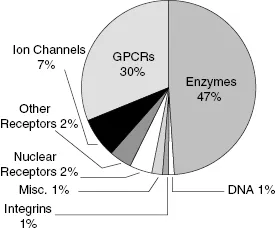
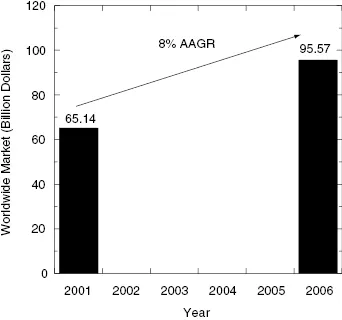
Enzymes, then, are attractive targets for drug therapy because of their essential roles in life processes and in pathophysiology. Indeed, a survey reported in 2000 found that close to 30% of all drugs in clinical use derive their therapeutic efficacy through enzyme inhibition (Drews, 2000). More recently Hopkins and Groom (2002) updated this survey to include newly launched drugs and found that nearly half (47%) of all marketed small molecule drugs inhibit enzymes as their molecular target (Figure 1.1). Worldwide sales of small molecule drugs that function as enzyme inhibitors exceeded 65 billion dollars in 2001, and this market was expected to grow to more than 95 billion dollars by 2006 (see Figure 1.2). Some contraction of the worldwide market has occurred due to withdrawal of several products since 2005. Revised forecasts suggest that the worldwide market will now grow at a rate of about 6.7% as of 2005 (Business Communications Company, Inc., 2006, “Enzyme Inhibitors with Broad Therapeutic Application”).
The attractiveness of enzymes as drug targets results not only from the essentiality of their catalytic activity but also from the fact that enzymes, by their very nature, are highly amenable to inhibition by small molecular weight, drug-like molecules. Because of this susceptibility to inhibition by small molecule drugs, enzymes are commonly the target of new drug discovery and design efforts at major pharmaceutical and biotechnology companies today; my own informal survey suggests that between 50 and 75% of all new drug-seeking efforts at several major pharmaceutical companies in the United States are focused on enzymes as primary targets.
While the initial excitement generated by the completion of the Human Genome Project was in part due to the promise of a bounty of new targets for drug therapy, it is now apparent that only a portion of the some 30,000 proteins encoded for by the human genome are likely to be amenable to small molecule drug intervention. A recent study suggested that the size of the human “druggable genome” (e.g., human genes encoding proteins that are expected to contain functionally necessary binding pockets with appropriate structures for interactions with drug-like molecules) is more on the order of 3000 target proteins (i.e., about 10% of the genome), a significant portion of these being enzymes (Hopkins and Groom, 2002). As pointed out by Hopkins and Groom, just because a protein contains a druggable binding pocket does not necessarily make it a good target for drug discovery; there must be some expectation that the protein plays some pathogenic role in disease so that inhibition of the protein will lead to a disease modification. Furthermore the same study estimates that of the nearly 30,000 proteins encoded by the human genome, only about 10% (3000) can be classified as “disease-modifying genes” (e.g., genes that, when knocked out in mice, effect a disease-related phenotype). The intersection of the druggable genome and the disease-modifying genome thus defines the number of bona fide drug targets of greatest interest to pharmaceutical scientists. This intersection, according to Hopkins and Groom (2002), contains only between 600 and 1500 genes, again with a large proportion of these genes encoding for enzyme targets.
The “druggability” of enzymes as targets reflects the evolution of enzyme structure to efficiently perform catalysis of chemical reactions, as discussed in the following section.