![]()
Chapter 1
Understanding Gene Duplication Through Biochemistry and Population Genetics
David A. Liberles and Grigory Kolesov
Department of Molecular Biology, University of Wyoming, Laramie, Wyoming
Katharina Dittmar
Department of Biological Sciences, SUNY at Buffalo, Buffalo, New York
1.1 Introduction
Gene duplication has emerged as an important process supporting the functional diversification of genes. Since publication of the seminal book Evolution by Gene Duplication by Ohno (1970), the hypothesis regarding the importance of gene duplication in the generation of evolutionary novelty has steadily gained support as we have entered the genome-sequencing era. It is through the link to functional biology that an ultimate understanding of the preservation and diversification of duplicate genes will be accomplished.
Genes can diverge in function through accumulation (fixation) of coding sequence changes, which may influence binding interactions and/or catalysis, through the evolution of splice variants, and through spatial, temporal, and concentration-level changes in the expression of the protein product. Governing these processes is an interplay among mutational opportunity, population dynamics, protein biochemistry, and systems and organismal biology. This interplay is described systematically in this chapter.
1.2 Systems Biology and Higher-Level Organization
At the level of biological systems, two early but still relevant views suggested a role for gene duplication in constructing pathways. These views are both dependent on a new function emerging in one of the duplicates, but differ in the manner in which it occurs. One view, patchwork evolution, involved a conservation of catalytic activity coupled with the evolution of a new substrate after duplication (Jensen, 1976). An alternative view, retrograde evolution, suggested that pathways are built up backward, with product becoming substrate based on recognition of the transition state in the active site, with the evolution of a new catalytic activity to generate the substrate for the downstream reaction after duplication (Horowitz, 1945). In a systematic analysis in Escherichia coli, Light and Kraulis found some evidence for the retrograde evolution model, but found the patchwork model to be much more common, possibly because it is easier to gain new binding specificity than to evolve a new catalytic activity (Light and Kraulis, 2004). Relatedly, it has been suggested that (also in bacteria) there are secondary (moonlighting) functions where enzymes with a given catalytic activity carry it out on multiple substrates with different specificities (Copley, 2003). This nature of enzymatic activities might generally lead to quick differential optimization after duplication, especially easily if maintained with different specificities in different alleles by balancing selection before duplication. Further (as discussed in detail below), specificity is chemically and evolutionarily difficult to attain, and nonspecific binding activities may arise easily when there is no selective pressure against them. Whereas selective pressures are ultimately at the systems level, divergence occurs gene by gene and mutation by mutation. This process will be dissected.
1.3 Mutational Dynamics and Substitutions
Both intramolecular and intermolecular coevolution of sites affects the probability of fixation of any individual mutation, where genetic background (the sequence at genetically interacting positions) determines the phenotype of any given mutation. The evolutionary accessibility of different mutations from a given genetic background is therefore dictated partly by the mutation rate and the frequency of multiple segregating mutations as well as the population size as a dictator of strength of selection. The same evolutionary properties affect both intramolecular and intermolecular interaction, only with differing degrees of sensitivity to mutation, due to the entropic differences between the two types of interresidue interaction. For these entropic reasons, it is easier to knock out a binding interaction than to knock out proper protein folding (although this happens, too) with a single mutation. This is because although there are a greater number of sites that influence proper folding, covalent attachment means that there will also be a greater local effective concentration of intramolecularly interacting residues requiring a lower affinity interaction to generate the same levels of bound state. If one views two residues as interacting or not interacting, the probability of interaction at any given time is dependent on their affinity for each other and how many opportunities they have to interact (their concentration about each other).
So far, we have focused on the coding properties of a gene. Gene expression is another important process that is subject to phenotypic divergence through mutation. The typical gene has approximately 12 transcription factor binding sites [the distribution of this across genomes is not well characterized, and this number is given with an approximation of six to eight base pairs (Harbison et al., 2004; Hughes and Liberles, 2007)]. The specificity of binding typically enables transcription factors to discriminate among many sites with single-base-pair mutations (Lusk and Eisen, 2008). Because of the small size of transcription factor–binding sites, site loss and de novo site evolution are reasonably common, and this is explored further below. Due to the periodicity of standard B-form DNA of about 10 bp, as well as changes in effective local concentration of transcription factors about each other and about the initiation site, it might be expected that spacing between sites is important in gene regulation, but evidence generated so far seems to downplay the role of these effects (Shultzaberger et al., 2007), leading to a focus on the evolution of the sites themselves.
Splicing is another mechanism by which genes can diverge through mutation. There are two types of splicing, constitutive and alternative, with alternative splicing simply showing a weaker consensus to splicing regulatory sites (Churbanov et al., 2008). Like transcription factor–binding sites, splicing regulatory sequences are short and potentially subject to turnover. However, because of the lack of redundancy (unlike transcription factor–binding sites), loss in the absence of duplication may frequently be highly deleterious. It has been shown that alternative splicing itself enables a substitution burst mediated by relaxed selection on and around these regulatory sites (Xing and Lee, 2005). That gene duplication can also enable such a burst of substitution under relaxed selection suggests that gene duplication should enable enhanced rates of alternative transcript generation, and this has indeed now been demonstrated (Jin et al., 2008).
Many other molecular mechanisms can contribute to mutation-driven diversification. A far from exhaustive list would include glycosylation sites, protein splicing, and RNA editing—one only needs to think of the effects of duplication and relaxed selection on any processes generating constraint described in a molecular biology textbook.
Starting with a few examples of several of these molecular processes, we will then link mutational opportunity to evolutionary mechanism and process. The following section includes a series of examples of the fates of duplicate genes. These examples are meant to be illustrative, and we will ultimately address how general the various processes that underlie the examples actually are.
1.4 Evolution of Enzyme Active Centers After Duplication
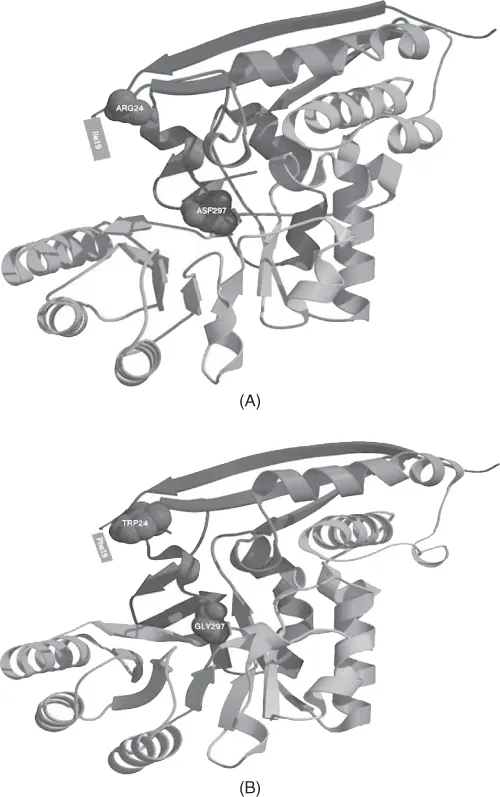
Mutations in the active center(s) of an enzyme can lead to a change of its substrate or a change in its kinetics. For example, Vick and Gerlt (2007) demonstrate that a single-base-pair change leading to D-to-G substitution in the active center of the monofunctional l-Ala-d/l-Glu epimerase from E. coli introduced the ability to catalyze the o-succinylbenzoate synthase reaction while reducing the level of the original reaction (Figure 1.1). Four additional nucleotide substitutions led to a complete switch of specificity and kinetics to the new reaction. Consistent with the patchwork model discussed earlier, a large number of enzymes in the arginine and lysine synthetic pathways are homologous to each other (Miyazaki et al., 2001).
Mutations in the structure surrounding the active center can lead to fine-tuning the active center to different but fundamentally similar substrates. For example, residues in the active centers of Leu-tRNA synthase and Ile-tRNA synthase are mostly conserved; Leu and Ile are very close chemically. There are a number of variable residues that do not directly contact the substrate residue in the active center but, rather, shape the active center, allowing for recognition of the cognate substrate residue. Both tRNA synthases are highly similar on both the sequence and structural levels. Leu- and Ile-tRNA synthases probably arose via gene duplication (Brown and Doolittle, 1995). This demonstrates a shift in substrate specificity following gene duplication.
1.4.1 Change of DNA-Binding Specificity
Homeobox genes are homeodomain-containing transcription factors that are known as principal regulators in the formation of the animal body plan during embryo development. They are often organized in homologous gene clusters such as Hox, ParaHox, and NK (Garcia-Fernández, 2005). Hox clusters can contain different numbers of genes in different species, where new genes in the clusters arise via duplication and loss in the course of evolution.
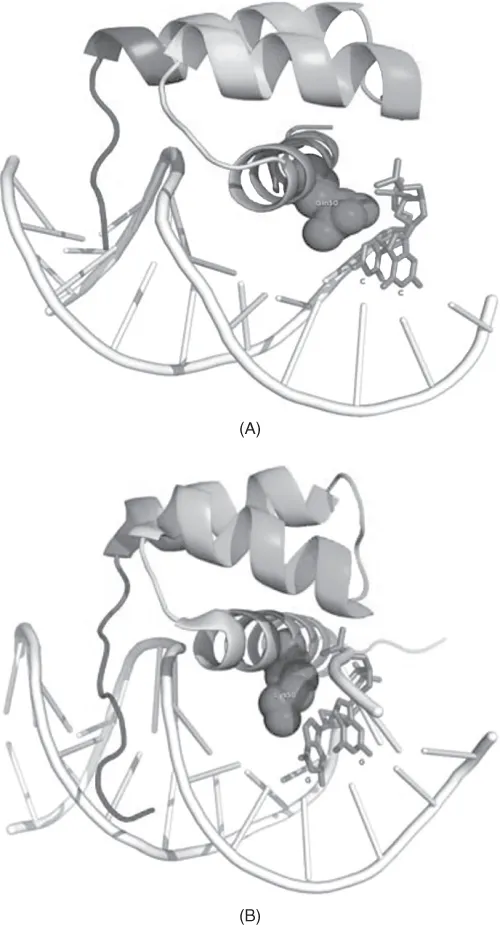
It has been shown that the DNA-binding specificity of Hox genes is controlled by a few key positions in the homeodomain. For example, substitution of Gln to Lys in position 50 of the homeodomain alters recognition from TAATCC (recognized by bicoid class hox proteins) to the TAAT(T/G)(A/G) motif recognized by the Antennapedia and Engrailed classes (Hanes and Brent, 1989; Treisman et al., 1989; Percival-Smith et al., 1990). This is shown in Figure 1.2. Similarly, substitutions in positions 3, 6, and 7 of the N-terminus of the homeodomain alter the specificity toward the nucleotide in position 2 of the motif TTATGG → TAATGG (Ekker et al., 1994; Noyes et al., 2008).
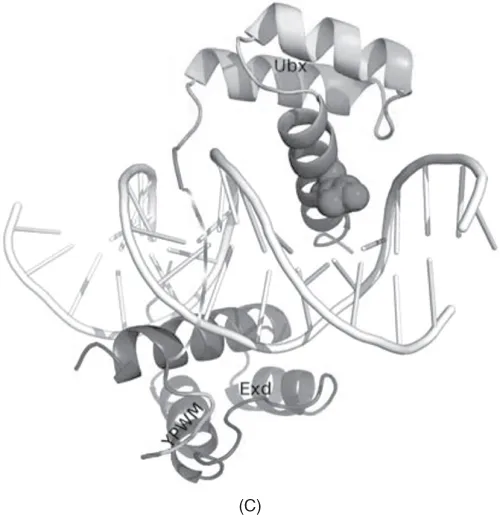
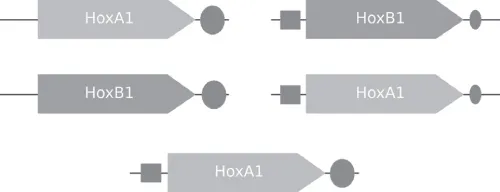
The evolution of homeotic genes in Hox-like clusters demonstrates how gene duplication followed by a single or a few mutations can create new functions that have dramatic effects on the phenotype (in the case of Hox genes, the number of body segments, limbs, etc.). An example of the rearrangement of Hox genes and their regulatory elements is shown in Figure 1.3.
1.4.2 Change of Binding Interface and Interaction Partners
Most proteins do not act alone but, instead, interact with other proteins. This is another mode of potential divergence for duplicated genes. Protein–protein interactions are in most cases highly specific and form complex protein interaction networks that execute metabolic functions and make up regulatory, signal transduction, and intercellular circuits. Mutations in the protein–prot...