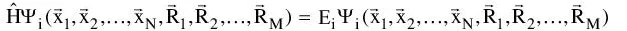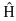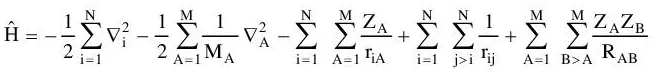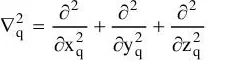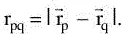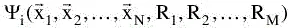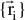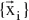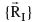"Chemists familiar with conventional quantum mechanics will applaud and benefit greatly from this particularly instructive, thorough and clearly written exposition of density functional theory: its basis, concepts, terms, implementation, and performance in diverse applications. Users of DFT for structure, energy, and molecular property computations, as well as reaction mechanism studies, are guided to the optimum choices of the most effective methods. Well done!" Paul von Rague Schleyer
"A conspicuous hole in the computational chemist's library is nicely filled by this book, which provides a wide-ranging and pragmatic view of the subject.[...It] should justifiably become the favorite text on the subject for practioneers who aim to use DFT to solve chemical problems." J. F. Stanton, J. Am. Chem. Soc.
"The authors' aim is to guide the chemist through basic theoretical and related technical aspects of DFT at an easy-to-understand theoretical level. They succeed admirably." P. C. H. Mitchell, Appl. Organomet. Chem.
"The authors have done an excellent service to the chemical community. [...] A Chemist's Guide to Density Functional Theory is exactly what the title suggests. It should be an invaluable source of insight and knowledge for many chemists using DFT approaches to solve chemical problems." M. Kaupp, Angew. Chem.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
What is density functional theory? The first part of this book is devoted to this question and we will try in the following seven chapters to give the reader a guided tour through the current state of the art of approximate density functional theory. We will try to lift some of the secrets veiling that magic black box, which, after being fed with only the charge density of a system somewhat miraculously cranks out its energy and other ground state properties. Density functional theory is rooted in quantum mechanics and we will therefore start by introducing or better refreshing some elementary concepts from basic molecular quantum mechanics, centered around the classical Hartree-Fock approximation. Since modern density functional theory is often discussed in relation to the Hartree-Fock model and the corresponding extensions to it, a solid appreciation of the related physics is a crucial ingredient for a deeper understanding of the things to come. We then comment on the very early contributions of Thomas and Fermi as well as Slater, who used the electron density as a basic variable more out of intuition than out of solid physical arguments. We go on and develop the red line that connects the seminal theorems of Hohenberg and Kohn through the realization of this concept by Kohn and Sham to the currently popular approximate exchange-correlation functionals. The concept of the exchange-correlation hole, which is rarely discussed in detail in standard quantum chemical textbooks holds a prominent place in our exposition. We believe that grasping its characteristics helps a lot in order to acquire a more pictorial and less abstract comprehension of the theory. This intellectual exercise is therefore well worth the effort. Next to the theory, which – according to our credo – we present in a down-to-earth like fashion without going into all the many intricacies which theoretical physicists make a living of, we devote a large fraction of this part to very practical aspects of density functional theory, such as basis sets, numerical integration techniques, etc. While it is neither possible nor desirable for the average user of density functional methods to apprehend all the technicalities inherent to the implementation of the theory, the reader should nevertheless become aware of some of the problems and develop a feeling of how a solution can be realized.
1 Elementary Quantum Chemistry
In this introductory chapter we will review some of the fundamental aspects of electronic structure theory in order to lay the foundations for the theoretical discussion on density functional theory (DFT) presented in later parts of this book. Our exposition of the material will be kept as brief as possible and for a deeper understanding the reader is encouraged to consult any modern textbook on molecular quantum chemistry, such as Szabo and Ostlund, 1982, McWeeny, 1992, Atkins and Friedman, 1997, or Jensen, 1999. After introducing the Schrödinger equation with the molecular Hamilton operator, important concepts such as the antisymmetry of the electronic wave function and the resulting Fermi correlation, the Slater determinant as a wave function for non-interacting fermions and the Hartree-Fock approximation are presented. The exchange and correlation energies as emerging from the Hartree-Fock picture are defined, the concepts of dynamical and nondynamical electron correlation are discussed and the dissociating hydrogen molecule is introduced as a prototype example.
1.1 The Schrödinger Equation
The ultimate goal of most quantum chemical approaches is the – approximate – solution of the time-independent, non-relativistic Schrödinger equation
(1-1)
where
is the Hamilton operator for a molecular system consisting of M nuclei and N electrons in the absence of magnetic or electric fields.
is a differential operator representing the total energy:
(1-2)
Here, A and B run over the M nuclei while i and j denote the N electrons in the system. The first two terms describe the kinetic energy of the electrons and nuclei respectively, where the Laplacian operator
is defined as a sum of differential operators (in cartesian coordinates)
(1-3)
and MA is the mass of nucleus A in multiples of the mass of an electron (atomic units, see below). The remaining three terms define the potential part of the Hamiltonian and represent the attractive electrostatic interaction between the nuclei and the electrons and the repulsive potential due to the electron-electron and nucleus-nucleus interactions, respectively. rpq (and similarly Rpq) is the distance between the particles p and q, i. e.,
stands for the wave function of the i’th state of the system, which depends on the 3N spatial coordinates
, and the N spin coordinates1 {si} of the electrons, which are collectively termed
and the 3M spatial coordinates of the nuclei,
The wave function ψi contains all information that can possibly be known about the quantum system at hand. Finally, Ei is the numerical value of th...
Table of contents
Cover
Table of Contents
Series
Title
Copyright
Foreword
Preface
Preface to the second edition
PART A: The Definition of the Model
PART B: The Performance of the Model
Bibliography
Index
End User License Agreement
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
Both plans are available with monthly, semester, or annual billing cycles.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go. Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access A Chemist's Guide to Density Functional Theory by Wolfram Koch,Max C. Holthausen in PDF and/or ePUB format, as well as other popular books in Physical Sciences & Physical & Theoretical Chemistry. We have over 1.5 million books available in our catalogue for you to explore.