![]()
PART I
GROWTH FACTOR INHIBITORS: VEGFR 2, E RB B 2, AND OTHER KINASES
![]()
1
DISCOVERY AND DEVELOPMENT OF SUNITINIB (SU 11248): A MULTITARGET TYROSINE KINASE INHIBITOR OF TUMOR GROWTH, SURVIVAL, AND ANGIOGENESIS
CONNIE L. SUN, JAMES G. CHRISTENSEN, AND GERALD McMAHON
1.1. SUNITINIB
Sunitinib (SU11248, Sutent™; Pfizer, Inc.) is an oral, multitargeted tyrosine kinase inhibitor of vascular endothelial growth factor receptors (VEGFRs 1, 2, and 3), platelet-derived growth factor receptors (PDGFRs α and β), stem-cell factor receptor (KIT), FMS-like tyrosine kinase 3 (FLT3), colony-stimulating factor 1 receptor (CSF1R), and glial cell line-derived neurotrophic factor receptor (REarranged during Transfection; RET) and currently approved multinationally for the treatment of advanced renal cell carcinoma (RCC) and for gastrointestinal stromal tumors (GISTs) after disease progression or intolerance to imatinib mesylate (IM) therapy. Sunitinib (SU11248) emerged from a drug discovery program within the biotechnology company (SUGEN, South San Francisco). An original aim of the SU11248 program was to understand cellular signaling mechanisms and design novel agents that would modulate signaling mechanisms for the treatment of human disease. Sunitinib resulted from many years of iterative chemistry and pharmacologic testing. Herein, we summarize the historical discovery and rationale, clinical pharmacology, non-clinical and translational medicine studies supporting early clinical evaluation, and clinical trial data that led to product approval in 2006.
1.2. RECEPTOR TYROSINE KINASE SIGNALING AFFECTS MULTIPLE AND DIVERSE CANCER PROCESSES
In the mid-to-late 1980s through the early 1990s, advances in molecular biology and DNA cloning techniques resulted in the discovery and functional characterization of genes involved in the etiology and progression of human cancer. During this period the identification of oncogenes capable of transforming healthy cells to malignant cells provided new understanding of the encoded proteins critical to signal transduction pathways and the mechanisms promoting tumor progression. The comprehensive characterization of these oncoproteins identified protein tyrosine kinases and receptor tyrosine kinase (RTK) families (Manning et al., 2002).
RTK signaling is normally tightly regulated and is critically important in processes, such as cell proliferation and survival, migration, and metabolism (Schlessinger, 2000). To date, at least 60 RTK transmembrane signaling proteins have been identified. Each RTK is characterized by an extracellular domain that functions as a binding site for a specific polypeptide ligand, a cytoplasmic kinase catalytic domain, and additional regulatory sequences that facilitate downstream signal transduction. The stimulation of RTK catalytic activity in response to ligand binding initiates the transfer of ATP gamma -phosphate to tyrosine residues on target proteins. This phosphorylation initiates a downstream intracellular signaling cascade.
The dysregulation of RTK signaling caused by a mutation, ectopic receptor, or ligand expression has been implicated in aspects of tumor progression including cell proliferation, survival, angiogenesis, and tumor dissemination (Blume-Jensen and Hunter, 2001). The characterization and expression of tyrosine kinases at the RNA and protein levels indicated that these tyrosine kinases were often highly expressed in tumor tissue relative to normal or surrounding adjacent tissues. The early protein tyrosine kinase drug targets were mostly RTKs associated with epithelial-derived cancers. Drug discovery research efforts in human cancer revolved around these early drug targets such as the epidermal growth factor receptor (EGFR, erbB1, HER1), epidermal growth factor receptor 2 (HER2, erbB2, neu), fibroblast growth factor receptor 1 (FGFR1), VEGFR, PDGFR, insulin-like growth factor receptor-1 (IGF-R1), and c-src, a cytoplasmic membrane-associated tyrosine kinase.
1.3. ROLE OF RECEPTOR TYROSINE KINASES IN PATHOLOGIC TUMOR ANGIOGENESIS
It has long been known that neoplasms are often accompanied by an increased and unique vascularity; however, the molecular mechanisms responsible for these vascular changes in tumor tissue were not well understood and remained elusive for over 50 years (Lewis 1927; Tannock 1968). In 1971 Judah Folkman and colleagues introduced the concept of neoangiogenesis as a critical mechanism by which neoplasms secrete a “tumor angiogenic factor or TAF” in order to recruit vascular supply and gain access to nutrients and oxygen (Folkman et al., 1971). The field gained further, newfound attention in the mid-1980s through the early 1990s when a series of soluble factors and their receptors were implicated in endothelial cell function and angiogenesis. Some of the initial factors shown to regulate endothelial cell function, angio-genesis, and vascular permeability included peptide growth factors, such as vascular permeability factor (VPF) (Senger et al., 1983) also known as vascular endothelial growth factor (VEGF) (Ferrara and Henzel, 1989), basic and acidic fibroblast growth factors (bFGF and aFGF) (Maciag et al., 1984; Shing et al., 1984), angiogenin (Fett et al., 1985), and platelet-derived growth factor (PDGF) (Folkman, 1995). When vascular permeability factor (VPF) was first identified, studies by Senger and Dvorak showed that when partially purified as a protein factor it was able to induce vascular leakage (Senger et al., 1983). Ferrara and colleagues cloned a gene product that functioned as a diffusible, soluble, and selective endothelial cell mitogen, which they termed vascular endothelial growth factor (VEGF) (Ferrara and Henzel, 1989). Subsequent cloning and characterization by multiple laboratories and investigators suggested that this factor existed in several low molecular weight species (i.e., 121, 165, and 189 amino acids) and that VPF and VEGF were determined to be the same protein (Ferrara, 2004). Efforts to identify receptors for VEGF resulted in the cloning and identification of two receptor tyrosine kinases termed vascular endothelial growth factor receptor 1 (VEGFR1, FLT1) and factor 2 (VEGFR2, KDR) in 1992 (deVries et al., 1992; Terman et al., 1992). Both receptors could bind to VEGF with high affinity, were expressed predominantly in endothelial cells, and mediated its known biological effects in endothelial cell lines. When VEGF or VEGF receptor gene loci were disrupted in mice, findings suggested that these genes were required for mammalian development and for normal embryonic vascular development, thus providing evidence of their important role as key regulators of angiogenesis (Fong et al., 1995; Carmeliet et al., 1996; Ferrara et al., 1996).
Following the discovery and characterization of VEGFs and VEGFRs, a number of studies further supported the importance of VEGF and VEGFR in tumor angiogenesis. A group of studies in 1992 reported that VEGF was highly expressed in glioblastoma multiforme and that under hypoxic conditions, VEGF could be induced. This concept was found to be common to many tumor types (Shweiki et al., 1992; Plate et al., 1992). Subsequently, the overexpression of VEGF was noted in a variety of tumor types as well as identified as an independent prognostic factor (Folkman, 1995). Additional mechanistic studies demonstrated that stable transfection of nontumorigenic cell lines with VEGF allowed these cell lines to grow with vasculature as tumor xenografts in athymic mice despite having no effect on tumor growth properties in vitro (Claffey et al., 1996; Ferrara et al., 1993). Subsequent critical experiments were designed to address whether inhibiting the function of VEGF or VEGFRs could inhibit the progression of experimental tumors. The neutralization of soluble VEGF with monoclonal antibodies demonstrated the ability to inhibit tumor growth in a variety of tumor xenograft models despite having no effect on the growth properties of the respective cell lines in vitro (Kim et al., 1993). Similarly, dominant-negative Flk1 (the mouse ortholog of VEGFR2) retrovirus was used to inhibit Flk1/VEGFR2 activity and function in mouse endothelial cells in the setting of experimental tumor models. The Flk1 dominant-negative retrovirus inhibited angiogenesis and tumor growth in several experimental tumor models and represented early data that inhibition of VEGFR2 signaling may be a viable cancer treatment strategy (Millauer et al., 1994, 1996).
The discovery of multiple and distinct RTKs important in tumorigenesis and tumor angiogenesis prompted identification of monoclonal antibodies and specific chemicals that could inhibit protein function and cell signaling. Initial nonclinical data and proven clinical utility of therapeutic monoclonal antibodies, including trastuzamab, helped foster increased interest in RTK drug discovery. Similarly, initial nonclinical studies with VEGF-neutralizing antibodies helped build confidence in the targeting of this pathway to inhibit tumor angiogenesis and delay tumor progression. The identification of monoclonal antibodies that interrupt RTK signaling in animal cancer models has been reviewed elsewhere. With respect to chemical inhibition, initial approaches to identify chemicals that interrupted protein kinases began with natural product chemistry related to staurosporine and genistein and focused on enzyme catalytic function that could be purified from membrane preparations. These first chemicals from natural sources were found to be nonselective inhibitors of protein kinases but served as a starting point for novel synthetic chemistry. With the advent of DNA cloning and recombinant protein production, systematic drug design approaches were used to identify chemotypes that could interrupt protein kinase function. High-throughput screening and medicinal chemistry approaches using synthetic chemical templates such as the quinazolines, quinoxalines, and pyrimidines were the first to be evaluated. The initial protein kinase drug targets were Raf, p38, EGFR, PDGFR, and c-src protein kinases. In this regard, sunitinib evolved as the first small-molecule RTK inhibitor leading to the inhibition of tumor angiogenesis.
1.4. SU 5416: DISCOVERY OF FIRST-GENERATION VEGFR RTK INHIBITORS
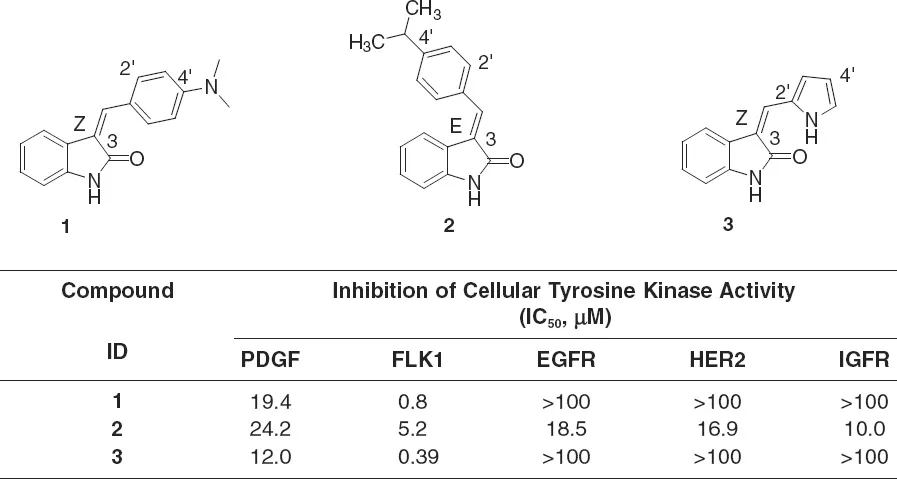
In 1994, the drug discovery program targeting tumor angiogenesis by inhibiting VEGFR catalytic activity was initiated at Sugen, Inc. A high-throughput screen identified the indolin-2-one chemotype with a biochemical PDGFR protein kinase assay. Among various hit chemotypes, indolin-2-ones demonstrated relative specificity in cells comparing VEGF to FGF signaling in cultured human umbilical vein endothelial cells (HUVECs) and comparing PDGF to EGF signaling in cultured fibroblasts. Three indolin-2-ones were identified as initial hits that exhibited inhibitory properties against various RTKs (1–3, Figure 1.1). Both 1 and 3 were found to be potent and selective inhibitors of VEGFR, whereas 2 was found to be nonselective for RTK inhibition.
Comparisons of these compounds suggested connections between chemical struc...