- Edited by prominent scientists working in drug discovery for Pfizer
- Meets the needs of a growing community of researchers in pharmaceutical R&D
- Provides a useful guide for practicing pharmaceutical scientists as well as a text for medicinal chemistry students
- An excellent follow-up to the very successful first book by these editors, Contemporary Drug Synthesis, but with all new therapeutic categories and drugs discussed.

eBook - ePub
The Art of Drug Synthesis
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
The Art of Drug Synthesis
About this book
The Art of Drug Synthesis illustrates how chemistry, biology, pharmacokinetics, and a host of other disciplines come together to produce successful medicines. The authors have compiled a collection of 21 representative categories of drugs, from which they have selected as examples many of the best-selling drugs on the market today. An introduction to each drug is provided, as well as background to the biology, pharmacology, pharmacokinetics, and drug metabolism, followed by a detailed account of the drug synthesis.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
CHAPTER 1
THE ROLE OF MEDICINAL CHEMISTRY IN DRUG DISCOVERY
1.1 INTRODUCTION
This volume represents the efforts of the many chemists whose ability to master both synthetic and medicinal chemistry enabled them to discover a new drug. Medicinal chemistry, like synthetic chemistry, comprises both art and science. It requires a comprehensive mind to collect and synthesize mountains of data, chemical and biological. It requires the instinct to select the right direction to pursue, and the intellect to plan and execute the strategy that leads to the desired compound. Most of all, it requires a balance of creativity and perseverance in the face of overwhelming odds to reach the goal that very few achieve—a successfully marketed drug.
The tools of medicinal chemistry have changed dramatically over the past few decades, and continue to change today. Most medicinal chemists learn how to use these tools by trial and error once they enter the pharmaceutical industry, a process that can take many years. Medicinal chemists continue to redefine their role in the drug discovery process, as the industry struggles to find a successful paradigm to fulfill the high expectations for delivering new drugs. But it is clear that however this new paradigm works out, synthetic and medicinal chemistry will continue to play a crucial role. As the chapters in this volume make clear, drugs must be successfully synthesized as the first step in their discovery. Medicinal chemistry consists of designing and synthesizing new compounds, followed by evaluation of biological testing results and generation of a new hypothesis as the basis for further compound design and synthesis. This chapter will discuss the role of both synthetic and medicinal chemistry in the drug discovery process in preparation for the chapters that follow on the syntheses of marketed drugs.
1.2 HURDLES IN THE DRUG DISCOVERY PROCESS
Although the tools of medicinal chemistry may have improved considerably (as discussed below), the hurdles to discovering a new drug have outpaced this improvement, accounting to a certain extent for the dearth of newly marketed drugs. Discussion of some of these hurdles, such as external pressures brought on by the public media and the stock market, lies outside the scope of this review. Instead, we will discuss those aspects of drug discovery under the control of the scientists involved.
One of the first challenges for the medicinal chemist assigned to a new project is to read the biology literature pertaining to its rationale. Interacting with biology colleagues and understanding the results from biological assays are critical to developing new hypotheses and program directions. Given the increasing complexity of current biological assays, more information is available, but incorporating it into chemistry planning requires more extensive biological understanding. This complexity applies to both the primary in vitro assay for the biological target thought to be linked to clinical efficacy, as well as selectivity assays for undesired off-target in vitro activities. Some of the same considerations apply to the increasingly sophisticated assays for other aspects of drug discovery, such as ADME (absorption, distribution, metabolism, and elimination) and safety, as summarized in Table 1.1.
TABLE 1.1. Important Considerations for the Medicinal Chemists
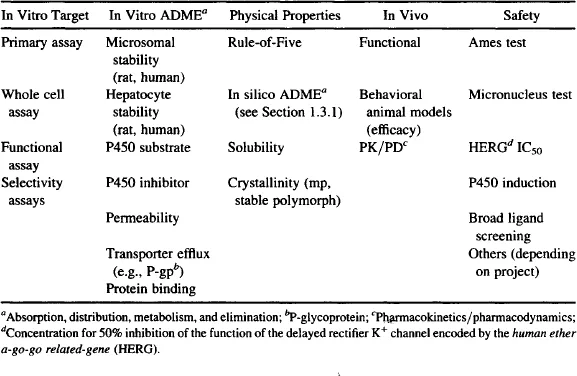
The reader is referred to an excellent overview of the biology behind these assays, and their deployment in a typical drug discovery program (Lin et al., 2003). The tools for addressing each of these hurdles fall into two categories, in silico modeling and structure-based drug design, which are covered in Sections 1.3.1 and 1.3.2. Obviously, the final hurdle is in vivo efficacy and safety data, which generally determine a compound’s suitability for advancement to clinical evaluation.
1.3 THE TOOLS OF MEDICINAL CHEMISTRY
1.3.1 In Silico Modeling
To overcome the many hurdles to discovering a new drug, medicinal chemists must focus on synthesizing compounds with drug-like properties. One of the first tools developed to help chemists design more drug-like molecules takes advantage of an area totally under the chemist’s control—the physical properties of the compounds being designed. These are the rules developed by Chris Lipinski, sometimes referred to as the “Rule-of-Five” (Ro5), which describe the attributes drug-like molecules generally possess that chemists should try to emulate (Lipinski et al., 2001). The Ro5 states that drug-like molecules tend to exhibit four important properties, each related to the number 5 (molecular weight <500; cLogP, a measure of lipophilicity,<5; H-bond donors <5; and H-bond acceptors <10). The Ro5 can be applied all the way from library design in the earliest stages of drug discovery to the final fine-tuning process that leads to the compound selected for development. Correlating microsomal instability and/or absorption/efflux with Ro5 properties can also provide insight about the property most important for gaining improvement in these areas.
As is the case with any good model, the Ro5 is based on data, in this case from hundreds of marketed drugs. Using more specific data, models to address each of the hurdles in the drug discovery process have been developed (for comprehensive reviews, see Beresford et al., 2004; van de Waterbeemd and Gifford, 2003; Winkler, 2004). These include models of solubility (Cheng and Merz, 2003; Hou et al., 2004; Liu and So, 2001), absorption/permeability (Bergstroem, 2005; Stenberg et al., 2002), oral bioavailability (Stoner et al., 2004), brain penetration (Abbott, 2004; Clark, 2003) and P450 interaction (de Graaf et al., 2005). More recently, the solution of X-ray crystal structures of the P450 enzymes 3A4 (Tickle et al., 2005) and 2D6 (Rowland et al., 2006) should enable application of structure-based drug design (see below) to help minimize interactions with these metabolic enzymes. Models for safety issues, such as genotoxicity (Snyder et al., 2004) and HERG (human ether a-go-go related-gene) interaction (which can lead to cardiovascular side effects due to QT prolongation) (Aronov, 2005; Vaz and Rampe, 2005) are also being developed. Although this profusion of in silico models offers considerable potential for overcoming hurdles in the drug discovery process, the models are only as good as the data used to build them, and often the best models are those built for a single project using data from only the compounds prepared for that specific project.
The models described above can be used, alone or in combination with structure-based drug design (see Section 1.3.2), to screen real or virtual libraries of compounds as an integral part of the design process. These improvements in library design, coupled with more efficient library synthesis and screening, provide value in both time and cost savings. The move towards using this library technology has been accelerated by the availability of a new resource for library generation: outsourcing (Goodnow, 2001). Contract research organizations (CROs) in the United States or offshore provide numerous synthetic services such as synthesis of literature standards, templates and monomers for library preparation, and synthesis of libraries (D’Ambra, 2003). These capabilities can relieve in-house medicinal chemists of much of the routine synthetic chemistry so they can focus on design and synthesis to enable new structure-activity relationships (SAR) directions. For an overview of the process as it fits together for the successful discovery of new drugs, see Lombardino and Lowe, 2004.
1.3.2 Structure-Based Drug Design (SBDD)
Progress in SBDD has been steady over the past two decades such that it has become a generally accepted strategy in medicinal chemistry, transforming the way medicinal chemists decide how to pursue their series’ SAR. Although obtaining X-ray crystallographic data for SBDD was achieved early on, it has taken many years to learn how to interpret, and not over-interpret, this data. Structural information on the protein target provided by X-ray crystallography offers the greatest structural resolution for docking proposed ligands, but other spectroscopic techniques, such as nuclear magnetic resonance (NMR), have demonstrated their utility as well. X-ray crystallography, however, is generally restricted to analysing soluble proteins such as enzymes. Also required is a ready source of large quantities of the target protein for crystallization, as is often the case for proteins obtained from microorganisms grown in culture.
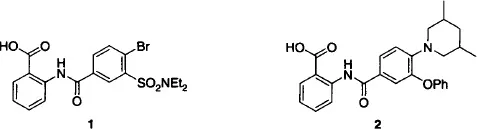
Bacterial proteins are an ideal starting point for SBDD, as in the case of the β-ketoacyl carrier protein synthase III (FabH), the target for a recent SBDD-based approach (Nie et al., 2005). FabH catalyzes the initiation of fatty acid biosynthesis, and a combination of X-ray data along with structures of substrates and known inhibitors led to selection of a screening library to provide a starting point for one recent study. Following screening, co-crystallization of selected inhibitors then guided the addition of functionality to take advantage of interactions with the enzyme visualized by X-ray and docking studies. A 50-fold improvement in enzyme inhibitory potency was realized in going from structure 1 to 2, accounted for by amino acid side-chain movements revealed by X-ray co-crystal structures of both compounds with the enzyme. Although much remains to be learned so that these side-chain movements can be predicted and exploited for new compound design, the study nonetheless provides a successful example of the implementation of SBDD in drug design.
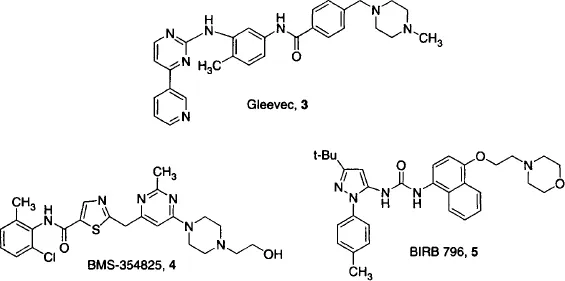
Although human proteins are more challenging to obtain in sufficient quantity for crystallization, modeling based on X-ray crystal structures has been successfully applied to many human targets. Probably the best-known efforts have been in the kinase area in search of anticancer drugs, which has been reviewed recently (Ghosh et al., 2001). For example. X-ray crystallographic data revealed important aspects of the binding of the anticancer drug Gleevec (3) to its target, the Bcr-Abl kinase, including the role of the pendant piperazine group, added originally to improve solubility, and the requirement for binding to an inactive conformation of the enzyme (Schindler et al., 2000). Combined with studies of the mutations responsible for Gleevec-resistant variants of Bcr-Abl, these studies enabled design of a new compound, BMS-354825 (4), active against most of these resistant mutants (Shah et al., 2004). More recently, non-ATP binding site inhibitors have been discovered and modeled by SBDD. For example, SBDD helped to characterize a new class of p38 kinase inhibitors that bind to a previously unobserved conformation of the enzyme that is incompatible with ATP binding (Pargellis et al., 2002). Insights from SBDD then guided design of a picomolar p38 kinase inhibitor based on binding to this site, BIRB 796 (5).
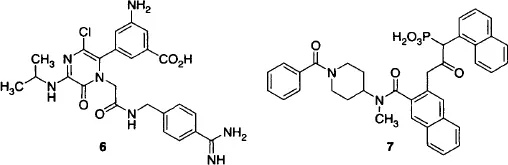
SBDD approaches to other soluble proteins have produced inhibitors of the tissue factor Vila complex (Parlow et al., 2003) and cathepsin G (Greco et al., 2002). In the case of factor Vila inhibitors, X-ray data provided information for both designing a new scaffold for inhibitors and for simultaneously improving binding affinity and selectivity over thrombin. Compound 6 from this work was advanced to clinical trials based on its potency and selectivity for factor Vila inhibition. The cathepsin G inhibitor program revealed a novel binding mode for an alpha-keto phosphonate to the enzyme’s oxyanion hole and active site lysine, as well as an opportunity to extend groups into a vacant binding site to improve potency. The result was a nearly 100-fold increase in inhibition following an SAR study of this direction using the amide group in compound 7.
Another spectroscopic technique that has been widely applied to drug design is nuclear magnetic resonance (N...
Table of contents
- Cover
- Half Title page
- Title page
- Copyright page
- Foreword
- Preface
- Contributors
- Chapter 1: The Role of Medicinal Chemistry in Drug Discovery
- Chapter 2: Process Research: How Much? How Soon?
- Part I: Cancer and Infectious Diseases
- Part II: Cardiovascular and Metabolic Diseases
- Part III: Central Nervous System Diseases
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access The Art of Drug Synthesis by Douglas S. Johnson, Jie Jack Li, Jie Jack Li,Douglas S. Johnson, Jie Jack Li, Douglas S. Johnson in PDF and/or ePUB format, as well as other popular books in Physical Sciences & Organic Chemistry. We have over 1.5 million books available in our catalogue for you to explore.