![]()
Part 1
Basic Principles
![]()
Chapter 1
Medical genetics in perspective
Key Topics
Scientific basis of medical genetics
Clinical applications of medical genetics
Introduction
Medical genetics is the science of human biological variation as it relates to health and disease. Although people have long been aware that individuals differ, that children tend to resemble their parents and that certain diseases tend to run in families, the scientific basis for these observations was only discovered during the past 140 years. The clinical applications of this knowledge are even more recent, with most progress confined to the past 50 years (see Table 1.1). In particular, the rapid sequencing of the entire human genome, completed in 2003, has greatly accelerated the process of gene mapping for genetic conditions and a vast quantity of valuable and continuously updated information has become readily accessible via the internet (as described in detail in Part 3 and on this book’s accompanying website at www.wiley.com/go/tobias).
Table 1.1 Some important landmarks in the development of medical genetics
| 1839 | Cell theory | Schleiden and Schwann |
| 1859 | Theory of evolution | Darwin |
| 1865 | Particulate inheritance | Mendel |
| 1882 | Chromosomes observed | Flemming |
| 1902 | Biochemical variation | Garrod |
| 1903 | Chromosomes carry genes | Sutton, Boveri |
| 1910 | First US genetic clinic | Davenport |
| 1911 | First human gene assignment | Wilson |
| 1944 | Role of DNA | Avery |
| 1953 | DNA structure | Watson, Crick, Franklin and Wilkins |
| 1956 | Amino acid sequence of sickle haemoglobin (HbS) | Ingram |
| 1956 | 46 chromosomes in humans | Tjio and Levan |
| 1959 | First human chromosomal abnormality | Lejeune |
| 1960 | Prenatal sexing | Riis and Fuchs |
| 1960 | Chromosome analysis on blood | Moorhead |
| 1961 | Biochemical screening | Guthrie |
| 1961 | X chromosome inactivation | Lyon |
| 1961 | Genetic code | Nirenberg |
| 1964 | Antenatal ultrasound | Donald |
| 1966 | First prenatal chromosomal analysis | Breg and Steel |
| 1966 | First print edition of Mendelian Inheritance in Man (MIM) | McKusick |
| 1967 | First autosomal assignment | Weiss and Green |
| 1970 | Prevention of Rhesus isoimmunisation | Clarke |
| 1970 | Chromosome banding | Caspersson and Zech |
| 1975 | DNA sequencing | Sanger, Maxam and Gilbert |
| 1976 | First DNA diagnosis | Kan |
| 1977 | First human gene cloned | Shine |
| 1977 | Somatostatin made by genetic engineering | Itakura |
| 1979 | In vitro fertilization | Edwards and Steptoe |
| 1979 | Insulin produced by genetic engineering | Goeddel |
| 1982 | First genetic engineering product marketed (Humulin) | Many contributors |
| 1985 | DNA fingerprinting | Jeffreys |
| 1986 | Polymerase chain reaction (PCR) | Mullis |
| 1987 | Linkage map of human chromosomes developed | Many contributors |
| 1987 | Online Mendelian Inheritance in Man (OMIM) first available | McKusick |
| 1990 | First treatment by supplementation gene therapy | Rosenberg, Anderson, Blaese |
| 1990 | First version of London Dysmorphology Database | Baraitser and Winter |
| 1990 | First clinical use of preimplantation genetic diagnosis (PGD) | Handyside, Winston and others |
| 1991 | First version of London Neurogenetics Database | Baraitser and Winter |
| 1993 | First physical map of the human genome | Many contributors |
| 2000 | First draft of the human genome sequence | Many contributors |
| 2003 | Completion of human genome sequencing (99.999%) | HGSC and Celera |
| 2006 | Preimplantation genetic haplotyping (PGH) announced | Renwick, Abbs and others |
| 2007 | Human genome SNP map (3.1 million SNPs) reported | International HapMap Consortium |
| 2007 | Completion of DNA sequencing of personal genomes | Watson and Venter |
| 2008 | Launch of project to sequence the genomes of over 1000 individuals from 20 different populations worldwide | International 1000 Genomes Project |
| 2010 | Publication of catalogue of human genetic variation (believed to be 95% complete) | International 1000 Genomes Project |
Scientific basis of medical genetics
Mendel’s contribution
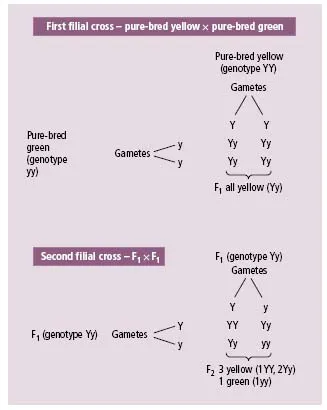
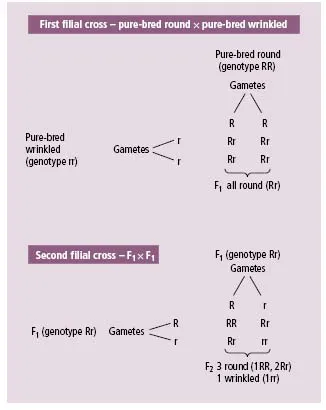
Prior to Mendel, parental characteristics were believed to blend in the offspring. While this was acceptable for continuous traits such as height or skin pigmentation, it was clearly difficult to account for the family patterns of discontinuous traits such as haemophilia or albinism. Mendel studied clearly defined pairs of contrasting characters in the offspring of the garden pea (Pisum sativum). These peas were, for example, either round or wrinkled and were either yellow or green. Pure-bred strains for each of these characteristics were available but when cross-bred (the first filial or F1 progeny) were all round or yellow. If F1 progeny were bred then each characteristic was re-observed in a ratio of approximately 3 round to 1 wrinkled or 3 yellow to 1 green (in the second filial or F2 progeny). Mendel concluded that inheritance of these characteristics must be particulate with pairs of hereditary elements (now called genes). In these two examples, one characteristic (or trait) was dominant to the other (i.e. all the F1 showed it). The fact that both characteristics were observed in the F2 progeny entailed segregation of each pair of genes with one member to one gamete and one to another gamete (Mendel’s first law).
Figures 1.1 and 1.2 illustrate these experiments with uppercase letters used for the dominant characteristic and lower-case letters used for the masked (or recessive) characteristic. If both members of the pair of genes are identical, this is termed homozygous (for the dominant or recessive trait), whereas a heterozygote has one gene of each type.
In his next series of experiments Mendel crossed pure-bred strains with two characteristics, e.g. pure-bred round/yellow with pure-bred wrinkled/green. The F1 generation showed only the two dominant characteristics – in this case round/yellow. The F2 showed four combinations: the original two, namely round/yellow and wrinkled/green, in a ratio of approximately 9:1 and two new combinations – wrinkled/yellow and round/ green in a ratio of approximately 3:3 (Fig. 1.3).
In these experiments, there was thus no tendency for the genes arising from one parent to stay together in the offspring. In other words, members of different gene pairs assort to gametes independently of one another (Mendel’s second law).
Although Mendel presented and published his work in 1865, after cultivating and studying around 28,000 pea plants, the significance of his discoveries was not realised until the early 1900s when three plant breeders, De Vries, Correns and Tschermak, confirmed his findings.
Chromosomal basis of inheritance
In 1839, Schleiden and Schwann established the concept of cells as the fundamental living units. Hereditary transmission through the sperm and egg was known by 1860, and in 1868, Haeckel, noting that the sperm was largely nuclear material, postulated that the nucleus was responsible for heredity. Flemming identified chromosomes within the nucleus in 1882, and in 1903 Sutton and Boveri independently realised that the behaviour of chromosomes during the production of gametes paralleled the behaviour of Mendel’s hereditary elements. Thus, the chromosomes were discovered to carry the genes. However, at that time, although the chromosomes were known to consist of protein and nucleic acid, it was not clear which component was the hereditary material.
Chemical basis of inheritance
Pneumococci are of two genetically distinct strains: rough or non-encapsulated (non-virulent) and smooth or encapsulated (virulent). In 1928, Griffith added heat-killed smooth bacteria to live rough bacteria and found that some of the rough pneumococci were transformed to the smooth, virulent type. Avery, MacLeod and McCarty repeated this experiment in 1944 and showed that nucleic acid was the transforming agent. Thus, nucleic acid was shown to carry hereditary information. This stimulated intense interest in the composition of nucleic acids, which culminated in the discovery, by Watson, Crick, Franklin and Wilkins, of the double-helical structure for deoxyribonucleic acid (DNA) in 1953.
Chromosomal disorders
By 1890, it was known that one human chromosome (the X chromosome) did not always have a partner, and in 1905 Wilson and Stevens extended this observation by establishing the pattern of human sex chromosomes. At this time, it was believed that there were 47 chromosomes, including one X chromosome, in each male somatic cell and 48 chromosomes, including two X chromosomes, in each female cell. In 1923, the small Y chromosome was identified, and both sexes were thought to have 48 chromosomes. Tjio and Levan refuted this in 1956 when they showed the normal human chromosome number to be 46. In 1959, the first chromosomal disease in humans, trisomy 21, was discovered by Lejeune and colleagues, and by 1970, over 20 different human chromosomal disorders were known. The development of chromosomal banding in 1970 markedly increased the ability to resolve small chromosomal aberrations, and so by 1990 more than 600 different chromosome abnormalities had been described, in addition to many normal variants. This number has increased further with the development of improved techniques including various fluorescence in situ hybridisation (FISH) methods and comparative genomic hybridisation (CGH). In fact, the increased resolution of the more recently developed techniques such as array CGH (see Chapter 7), has led to greater difficulties in differentiating between the increasingly numerous normal and abnormal chromosomal variants. This, in turn, has necessitated the development of international databases of such submicroscopic variants such as DECIPHER (Fig. 1.4), based at the Sanger Institute (http://decipher.sanger.ac.uk/), and the Database of Genomic Variants at Toronto (http://projects.tcag.ca/variation).
Mitochondrial disorders
Mitochondria have their own chromosomes and these are passed on from a mother to all of her children but not from the father. These chromosomes are different in several respects from their nuclear counterparts. For instance, they contain only 37 genes, a high and variable number of DNA copies per cell, very little non-coding DNA and no introns (see Chapter 5). Mutations in genes on these mitochondrial chromosomes can cause disease and this was first shown in 1988 for a maternally inherited type of blindness (Leber optic neuropathy). Since then, it has been shown that many different mitochondrial mutations, including point mutations, deletions and duplications, alone or in combination, can result in a variety of different disorders. Moreover, the relationship between genotype and phenotype is not straightforward, in part due to heteroplasmy, the tendency for a mitochondrial mutation to be present in only a proportion of the cell’s mitochondrial genome copies (see Chapter 10).
Single-gene disorders
In 1902, Garrod presented his studies on alkaptonuria, a rare condition in which patients have urine that darkens on standing and arthritis. He found three of 11 sets of parents of affected patients to be blood relatives and, in collaboration with Bateson, proposed that this was a Mendelian recessive trait with affected persons homozygous for the underactive gene. This was the first disease to be interpreted as a single-gene trait. Garrod also conceived the idea that patients with alkaptonuria and other inborn errors of metabolism really represented one extreme of human biochemical variation and that other less clinically significant variations were to be expected.
There followed numerous descriptions of distinct human single-gene traits and at the present time more than 7,000 human single-gene traits are known (Table 1.2). In 1949, Pauling suspected an abnormal haemoglobin to be the cause of sickle-cell anaemia, and this was confirmed by Ingram in 1956, who found an altered haemoglobin polypeptide sequence. This was the first demonstration in any organism that a mutation in a structural gene could produce an altered amino acid sequence. In 1959, only two abnormal haemoglobins were known; now the number exceeds 450. In 1948, Gibson demonstrated the first enzyme defect in an autosomal recessive condition (NADH-dependent methaemoglobin reductase in methaemoglobinaemia). The specific biochemical abnormalities in over 400 inborn errors of metabolism have now been determined, but the polypeptide product is still unknown in many human single-gene disorders. Study of these rare, and not so r...