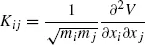This series provides the chemical physics field with a forum for critical, authoritative evaluations of advances in every area of the discipline. Volume 145 in the series continues to report recent advances with significant, up-to-date chapters by internationally recognized researchers.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Non-Markovian Theory of Vibrational Energy Relaxation and its Applications to Biomolecular Systems
Hiroshi Fujisaki,1,2 Yong Zhang,3 and John E. Straub4
1Molecular Scale Team, Integrated Simulation of Living Matter Group, Computational Science Research Program, RIKEN, 2-1 Hirosawa, Wako-shiSaitama 351-0198, Japan
2Department of Physics, Nippon Medical School 2-297-2 Kosugi-cho, Nakahara, Kawasaki, Kanagawa 211-0063, Japan
3Department of Chemical and Biomolecular Engineering, University of Notre Dame, 182 Fitzpatrick Hall, Notre Dame, IN 46556-5637, USA
4Department of Chemistry, oston University, 590 Commonwealth Avenue, SCI 503, Boston, MA 02215, USA
I. Introduction
Energy transfer (relaxation) phenomena are ubiquitous in nature. At a macroscopic level, the phenomenological theory of heat (Fourier law) successfully describes heat transfer and energy flow. However, its microscopic origin is still under debate. This is because the phenomena can contain many-body, multiscale, nonequilibrium, and even quantum mechanical aspects, which present significant challenges to theories addressing energy transfer phenomena in physics, chemistry, and biology [1]. For example, heat generation and transfer in nanodevices is a critical problem in the design of nanotechnology. In molecular physics, it is well known that vibrational energy relaxation (VER) is an essential aspect of any quantitative description of chemical reactions [2]. In the celebrated RRKM theory of an absolute reaction rate for isolated molecules, it is assumed that the intramolecular vibrational energy relaxation (IVR) is much faster than the reaction itself. Under certain statistical assumptions, the reaction rate can be derived [3]. For chemical reactions in solutions, the transition state theory and its extension such as Kramer's theory and the Grote–Hynes theory have been developed [4, 5] and applied to a variety of chemical systems including biomolecular systems [6]. However, one cannot always assume separation of timescales. It has been shown that a conformational transition (or reaction) rate can be modulated by the IVR rate [7]. As this brief survey demonstrates, a detailed understanding of IVR or VER is essential to study the chemical reaction and conformation change of molecules.
A relatively well-understood class of VER is a single vibrational mode embedded in (vibrational) bath modes. If the coupling between the system and the bath modes is weak (or assumed to be weak), a Fermi's-golden-rule style formula derived using second-order perturbation theory [8–10] may be used to estimate the VER rate. However, the application of such theories to real molecular systems poses several (technical) challenges, including how to choose force fields, how to separate quantum and classical degrees of freedom, or how to treat the separation of timescales between system and bath modes. Multiple solutions have been proposed to meet those challenges leading to a variety of theoretical approaches to the treatment of VER [11–16]. These works using Fermi's golden rule are based on quantum mechanics and are suitable for the description of high-frequency modes (more than thermal energy
200 cm−1), on which nonlinear spectroscopy has recently focused [17–20].
In this chapter, we summarize our recent work on VER of high-frequency modes in biomolecular systems. In our previous work, we have concentrated on the VER rate and mechanisms for proteins [21]. Here we shall focus on the time course of the VER dynamics. We extend our previous Markovian theory of VER to a non-Markovian theory applicable to a broader range of chemical systems [22, 23]. Recent time-resolved spectroscopy can detect the time course of VER dynamics (with femtosecond resolution), which may not be accurately described by a single timescale. We derive new formulas for VER dynamics and apply them to several interesting cases, where comparison to experimental data is available.
This chapter is organized as follows: In Section II, we briefly summarize the normal mode concepts in protein dynamics simulations, on which we build our non-Markovian VER theory. In Section III, we derive VER formulas under several assumptions and discuss the limitations of our formulas. In Section IV, we apply the VER formulas to several situations: the amide I modes in isolated and solvated N-methylacetamide and cytochrome c, and two in-plane modes (ν4 and ν7 modes) in a porphyrin ligated to imidazole. We employ a number of approximations in describing the potential energy surface (PES) on which the dynamics takes place, including the empirical CHARMM [24] force-field and density functional calculations [25] for the small parts of the system (N-methylacetamide and porphyrin). We compare our theoretical results with experiment when available, and find good agreement. We can deduce the VER mechanism based on our theory for each case. In Section V, we summarize and discuss the further aspects of VER in biomolecules and in nanotechnology (molecular devices).
II. Normal Mode Concepts Applied to Protein Dynamics
Normal mode provides a powerful tool in exploring molecular vibrational dynamics [26] and may be applied to biomolecules as well [27]. The first normal mode calculations for a protein were performed for BPTI protein [28]. Most biomolecular simulation softwares support the calculation of normal modes [24, 29, 30]. However, the calculation of a mass-weighted Hessian Kij, which requires the second derivatives of the potential energy surface, with elements defined as
(1)
can be co...
Table of contents
Cover
Editorial Board
Title Page
Copyright
Contributors to Volume 145
Introduction
Preface
Chapter 1: Non-Markovian Theory of Vibrational Energy Relaxation and its Applications to Biomolecular Systems
Chapter 2: Protein Functional Motions: Basic Concepts and Computational Methodologies
Chapter 3: Non-Brownian Phase Space Dynamics of Molecules, the Nature of Their Vibrational States, and Non-RRKM Kinetics
Chapter 4: Dynamical Reaction Theory Based on Geometric Structures in Phase Space
Chapter 5: Ergodic Problems for Real Complex Systems in Chemical Physics
Subject Index
Author Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
Both plans are available with monthly, semester, or annual billing cycles.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go. Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Advancing Theory for Kinetics and Dynamics of Complex, Many-Dimensional Systems by Tamiki Komatsuzaki, R. Stephen Berry, David M. Leitner, Tamiki Komatsuzaki,R. Stephen Berry,David M. Leitner, Stuart A. Rice,Aaron R. Dinner in PDF and/or ePUB format, as well as other popular books in Physical Sciences & Physical & Theoretical Chemistry. We have over 1.5 million books available in our catalogue for you to explore.