
eBook - ePub
Soft Tissue Tumors
A Multidisciplinary, Decisional Diagnostic Approach
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
About this book
Soft tissue tumors (STTs) are frequently misdiagnosed in inexperienced hands. Having diagnosed and treated hundreds of patients with these difficult tumors in the last few years, Institut Curie physicians have collected core data contributing to breakthrough research into the morphological, biological, and molecular aspects of soft tissue tumors, resulting in valuable translational and clinical applications to patient treatment. Soft Tissue Tumors: A Multidisciplinary, Decisional Diagnostic Approach presents a distillation of these experiences, combined with valuable data and perspectives contributed by senior pathologists, oncologists, and radiologists from several of the world's other leading cancer centers of excellence.
Information
Chapter 1
Clinical Approach in Soft Tissue Tumors
1.1 EPIDEMIOLOGY
Sarcomas are rare malignant tumors that originate from mesenchymal tissue at any body site. Soft tissue sarcomas comprise approximately 1% of malignant tumors [1,2]. There are more than 50 subtypes, pleomorphic sarcoma, liposarcoma, leiomyosarcoma, synovial sarcoma, and malignant peripheral nerve sheath tumor accounting for 75% of the cases. Roughly speaking, 80% of sarcomas originate from soft tissues, whereas the remainder are from bones. More than 10,000 new cases are diagnosed each year in the United States [1,2]. They account for 0.72% of all cancers diagnosed annually, whereas they represent 7% of all cancers in children. In Europe, similar data are reported with almost 8% of neoplasms in children, and almost half of them being less than 5 years of age at diagnosis [3]. Between 1988 and 1997, the age-standardized incidence of soft tissue sarcomas in Europe was 9.1 per million children, with a lower range of affected patients in the western and eastern parts of the continent and a higher one in the northern. The annual incidence is 30 per million [3].
Most soft tissue sarcomas occur in adults older than 55 years. Approximately 50% of bone sarcomas and 20% of soft tissue sarcomas are diagnosed in people younger than 35 years. The gastrointestinal stromal tumor (GIST), the frequency of which has been underestimated, is the most common form of soft tissue neoplasm. Its incidence, prevalence, and clinical aggressiveness also have been underestimated [4]. More recent experience from epidemiologic studies and active GIST therapeutic trials suggest that the annual incidence of GIST in the United States is at least 4000 to 6000 new cases (roughly 7 to 20 cases per million population per year) [4]. Some sarcomas, such as leiomyosarcoma, chondrosarcoma, and GIST are more common in adults than in children. Only approximately 500 thigh liposarcomas are diagnosed per year in the United States compared with more than 212,900 adenocarcinomas of the female breast. Thus, the number of adenocarcinomas originating in one anatomic site in women exceeds by almost 500-fold the number of thigh liposarcomas and is 22-fold higher than the total number of soft tissue sarcomas of all pathological varieties, at all anatomical sites, in all age groups, and in both genders.
Most high-grade bone sarcomas, including Ewing sarcoma/peripheral neuroectodermal tumor and osteosarcoma, are much more common in children and young adults. Among children, soft tissue sarcomas are two times more common in Caucasians than in African Americans. Rhabdomyosarcoma is the most frequent childhood soft tissue sarcoma (50%). Population-based data from Connecticut covering the years 1935–1989 have shown an increased incidence of soft tissue sarcomas in both genders, with men being more affected than women. The recent increase of acquired immune deficiency syndrome–related Kaposi sarcoma does not explain the upward trend in soft tissue sarcoma, dating back decades. A similar trend was found in a population-based study including 5802 cases of soft tissue sarcomas in children aged 0–14 years, which was extracted from the database of the Automated Childhood Cancer Information System (ACCIS) and registered in population-based cancer registries in Europe for the period 1978–1997. The incidence of soft tissue sarcomas in children increased by almost 2% per year during the period 1978–1997 as a result of the higher incidence of genitourinary rhabdomyosarcoma [3]. In most cases of soft tissue sarcomas, precise etiology is unknown, although several associated or predisposing factors have been identified, including environmental, physical, biological, and chemical factors.
1.1.1 Previous Local Injury
Soft tissue sarcomas can develop in areas of scar tissue after surgery, burns, fractures, radiation therapy [5–10], chronic irritation, and lymphedema out. The number of cancer patients who live longer after a curative treatment of a primary neoplasm is increasing. Therefore, childhood cancer survivors have an increased risk for developing secondary sarcomas. Postirradiation sarcoma, although uncommon, is more frequent because the number of long-term survivors increases [5]. The risk of subsequent bone cancer among 9170 patients who had survived 2 or more years after the diagnosis of a cancer in childhood has been estimated [6]. As compared with the general population, the patients had a relative risk of 133 (95% confidence interval [CI], 98–176) and a mean ± (standard error [SE]) 20-year cumulative risk of 2.8 ± 0.7% [6]. A large cohort of childhood cancer survivors was followed to determine the true incidence of secondary sarcomas. The history of secondary sarcomas in 14,372 participants in the Childhood Cancer Survivor Study was determined from self-reports in three questionnaires [7]. A total of 108 patients developed sarcomas in a median of 11 years after the initial diagnosis of childhood cancer. The risk of sarcoma was more than nine-fold higher among childhood cancer survivors than among the general population (standardized incidence ratio [SIR] = 9.02, 95% CI = 7.44–10.93). The excess absolute risk of secondary sarcoma was 32.5 per 100,000 person-years (95% CI = 26.1–40.3 per 100,000 person-years). Higher standardized incidence ratios and excess absolute risks were associated with a young age at primary diagnosis, a primary sarcoma diagnosis, and a family history of cancer. In a multivariable model, an increased risk of secondary sarcoma was associated with radiation therapy (relative risk [RR] = 3.1, 95% CI = 1.5–6.2), a primary diagnosis of sarcoma (RR = 10.1, 95% CI = 4.7–21.8), a history of other secondary neoplasms (RR = 2.2, 95% CI = 1.1–4.5), and treatment with higher doses of anthracyclines (RR = 2.3, 95% CI = 1.2–4.3) or alkylating agents (RR = 2.2, 95% CI = 1.1–4.6) [6]. A study attempted to evaluate the risk of soft tissue sarcoma in areas close to previously irradiated anatomic regions in women with breast carcinoma. This population-based, retrospective cohort study allowed identifying 194,798 women who were diagnosed with invasive breast carcinoma between 1973 and 1995. According to data from the Surveillance, Epidemiology and End Results Program (SEER), 54 women in the radiation therapy cohort and 81 women in the nonradiation therapy cohort subsequently developed soft tissue sarcomas. In the radiation therapy cohort, the age-standardized incidence ratios were 26.2 (95% CI=16.5–41.4) for angiosarcoma and 2.5 (95% CI=1.8–3.5) for other sarcomas. In the nonradiation therapy cohort, the age-standardized incidence ratios were 2.1 (95% CI=1.0–4.4) and 1.3 (95% CI=1.0–1.7), respectively. The radiation therapy cohort demonstrated a greater risk for developing both angiosarcoma (RR=15.9, 95% CI=6.6–38.1) (Fig 1.1) and other sarcomas (RR=2.2, 95% CI=1.4–3.3) compared with the nonradiation therapy cohort, and the largest increase was observed in the chest wall/breast. The elevated RR was significant even within 5 years of radiation therapy but reached a maximum between 5 and 10 years. That study also showed that the risk of developing soft tissue sarcoma, especially angiosarcoma, was elevated after radiation therapy in women with breast carcinoma [8]. Eighty patients had a confirmed histologic diagnosis of sarcoma that occurred after radiation therapy during 1975 and 1995. The patients were treated for breast cancer (n = 33, 42%), non-Hodgkin lymphoma (n = 9, 11%), cervical cancer (n = 9, 11%), benign lesions (n = 4, 5%), or other tumors (n = 25, 31%). Sarcoma occurred after a mean latency of 12 years (range, 3–64 years), with most (70%) developing in the soft tissues [9]. In another study, the median dose of radiation delivered to the primary tumor site was 45 Gy, and the median interval between radiotherapy and a diagnosis of sarcoma was 14 years. Seven tumors were located in the anatomical region of the sternum, three were located on the lateral chest wall, and five were located in the thoracic outlet [10].
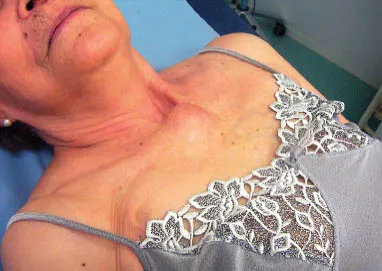
FIG 1.1 Presternal synovial sarcoma.
1.1.2 Exposure to Chemicals
The risk of developing a soft tissue sarcoma increases in patients who have been exposed to carcinogenic agents, particularly polycyclic hydrocarbons, asbestos, dioxin, and vinyl chloride [11–16]. The strongest documented association is related to vinyl chloride. In a case-control study of childhood rhabdomyosarcoma, families of 33 cases and 99 controls were interviewed. An RR of 3.9 was associated with fathers’ (but not mothers’) cigarette smoking (p =.003). For other cases, children had fewer immunizations than controls, particularly smallpox vaccinations (RR = 0.2; p =.001), and conversely had more preventive infections. An RR of 3.2 (p = .03) was found with exposure to chemicals, as well as with diets including giblets meats (RR of 3.7; p = .004). Mothers of affected children older than 30 years of age at the subject’s birth, those to be overaged at childbirth, and the role of antibiotics treatment preceding or during pregnancy also have been assessed. Other findings suggest that low socioeconomic status could be associated with an increased risk of rhabdomyosarcoma. All these findings suggest that environmental factors could play an important role in the etiology of childhood rhabdomyosarcoma [11]. Marijuana and cocaine addiction of parents during the year preceding their child’s birth has been reported to increase by two-fold to five-fold the risk of rhabdomyosarcoma in their children [12]. Exposure to phenoxyacetic acids has been associated with a roughly three-fold increased risk for soft tissue sarcoma, therefore confirming previous findings, whereas exposure to chlorophenols was not associated with a risk of developing soft tissue sarcomas in this study [13]. The potential role of phenoxy herbicides and chlorophenols in the development of soft tissue sarcomas also has been evaluated. In studies based on population referents, increased risks for soft tissue sarcoma were documented in gardeners (odds ratio [OR] = 4.1), railroad workers (OR = 3.1), as well as construction workers exposed to impregnating agents (OR = 2.3) [14]. Moreover, it also has been demonstrated [15] that soft tissue sarcoma risk, modeled using conditional logistic regression, was associated significantly with high-intensity chlorophenol exposure (OR = 1.79, 95% CI 1.10–2.88). A duration-response trend was evident among more highly exposed subjects (p < .0001). For subjects with 10 or more years of substantial exposure, the odds ratio was 7.78 (95% CI 2.46–24.65). These results suggest that chlorophenol exposure, independent of phenoxyherbicides, may increase the risk of soft tissue sarcoma [16].
1.1.3 Diseases or Conditions
Patients with weakened immune defenses such as human immunodeficiency virus (HIV) infection, congenital (inborn) immune deficiency, or immunosuppressive therapy are at risk for developing soft tissue sarcomas. Kaposi sarcoma is linked to HIV infection. HIV and human herpesvirus 8 has been implicated in the pathogenesis of Kaposi sarcoma. Rare familial syndromes with soft tissue sarcomas sarcoma have been identified. A report from the Cancer Family Registry of the National Cancer Institute allowed retrieving 24 kindreds of a syndrome that includes sarcoma, breast carcinoma, and other neoplasms in young patients. Cancer developed in an autosomal dominant pattern in 151 blood relatives, 119 (79%) of whom were affected before 45 years of age. These young patients had 50 bone and soft tissue sarcomas of diverse histological subtypes and 28 breast cancers. Additional features of the syndrome included an increased incidence of brain tumor (14 cases), leukemia (9 cases), and adrenocortical carcinoma (4 cases) before 45 years of age. These neoplasms also accounted for 73% of the multiple primary cancers occurring in 15 family members. This description led to the discovery of the Li-Fraumeni syndrome, which is related to p53 germline mutations [17,18]. New germline mutations of the p53 gene are rare among patients with “sporadic” sarcoma, whereas they are more frequent in patients whose background includes either multiple primary cancers or a family history of cancer [17]. As many as 7% of children with soft tissue sarcomas have Li-Fraumeni syndrome. The p53 gene seems altered in at least one third of sarcoma patients. In another third of patients, the MDM2 gene is amplified, resulting in an inhibition of the p53. Soft tissue sarcomas are more frequent among patients with certain inherited conditions including retinoblastoma [19], Li-Fraumeni syndrome, Gardners’s syndrome, Werner’s syndrome, nevoid basal cell carcinoma syndrome, neurofibromatosis type 1, and some immunodeficiency syndromes. Indeed, these risk factors account for a minority of soft tissue sarcomas, hence, the need for more genetic and environmental investigations.
1.2 CLINICS AND CLINICAL PROFILES
In day-to-day practice, the suspicion/diagnosis of a soft tissue sarcoma is unusual because of the rarity of these neoplasms; most patients seeking medical advice for a soft tissue lump do have a benign neoplastic or non-neoplastic condition. Some clinical presentations should suggest a reference to a specialist (those with a mass larger than 5 cm, as well as pain, increased size, deep to fascia, or a recurrent mass after previous excision). The definitive tests for diagnosing sarcoma are imaging (ultrasound, X-rays, computed tomography [CT] scan, or magnetic resonance imaging [MRI] scan) and a biopsy. It is important that both imaging and biopsy samples should be performed by experienced radiologists and pathologists in the management of soft tissue lesions. Many benign lumps simulate sarcomas, and they are obviously much more frequent. The contribution of pathologists to the multidisciplinary team of sarcomas, management and in the quality control of sarcoma diagnosis is mandatory. The planning of the surgical procedure follows the histological diagnosis. However, some sarcomas will come to be revealed or diagnosed in unexpected situations (e.g., uterine sarcoma in specimens of hysterectomy or GIST in resected abdominal and gastrointestinal masses). Otherwise, surgery should be undertaken under the supervision of a sarcoma specialist multidisciplinary team, even when the surgeon is not a regular member of that team.
1.2.1 Natural History of Soft Tissue Sarcomas
Soft tissue sarcomas can affect any part of the body. The most frequent location is the lower limb, accounting for about half of the cases, although the abdominal space and the retroperitoneum also are affected. Ideally, a definitive diagnosis should be stated in terms of benignancy or malignancy before any other procedure takes place. An initial surgical removal/biopsy is not recommended to avoid the contamination of the tumor bed, particularly for those lumps suspicious of malignancy. It has been demonstrated that the adequacy of an initial surgical resection was an important factor of prognosis, either in terms of recurrence and/or metastasis. Histologic evaluation of the surgical margins is mandatory [20] for both high- and low-grade neoplasms, whether superficial or deep. Approximately 50% of sarcoma patients will suffer a local recurrence and/or metastasis. For most histologic subtypes of soft tissue sarcomas, the most predictive factor of distant metastatic disease is tumor grade [21, 22]. The metastatic potential of low-grade sarcomas is 5–10%, of intermediate-grade sarcomas is 25–30%, and of high-grade sarcomas is approximately 50–60%. Additional histologic features have been used to evaluate tumor grade such as necrosis, pleomorphism, and the number of mitoses per microscopic high power field (HPF). However, some soft tissue sarcomas do not respond to the usual grading criteria; for example, tumors of the Ewing sarcoma/peripheral neuroectodermal tumor family are all high-grade sarcomas, whereas alveolar soft part sarcoma and some well-differentiated synovial sarcomas, although depicting a very low mitotic index and a well-recognized tumor type, are unpredictable in their biologic behavior.
Soft tissue sarcomas of the extremities usually metastasize to the lungs (70% of patients), with liver metastases being rare (<5%). Retroperitoneal and organ-based soft tissue sarcomas have a greater incidence of liver metastases with a similar rate of frequency to lung metastases. Myxoid liposarcoma is an exception, with a tendency to metastasize to other sites rather than to the lungs. Lymph node metastases are rare in soft tissue sarcomas and occur in less than 2–3% of cases, with the exception of synovial sarcoma, epithelioid sarcoma, and clear cell sarcoma of tendon sheath whose incidence of lymph node metastases reaches 20% [23].
Several low-grade soft tissue sarcomas are prone to gain a second clone of neoplastic cells during the course of their progression. These so-called “dedifferentiated sarcomas” depict a different phenotype of variable malignancy and can pursue a more aggressive clinical course [24–28]. In terms of oncogenesis, dedifferentiation is a phenomenon of tumor progression that seems to be time dependent. Dedifferentiation occurs in roughly 10% of well-differentiated liposarcomas of any subtype [25–28]. The risk of dedifferentiation is greater for deep-seated (particularly retroperitoneum) tumors and is significantly less for the limbs. Approximately 90% develop de novo, whereas 10% occur in recurrences. Dedifferentiation to leiomyosarcoma or rhabdomyosarcoma or less-differentiated liposarcoma is predictive of a worse prognosis in terms of recurrences, metastases, and survival. The use of microarray technology to evaluate gene expression profiles in biopsy or surgical specimens will provide newer insights into the pathogenesis of these tumors and therefore allow more precise subclassification and optimize the selection of therapeutic targets [8].
1.2.2 Age at Diagnosis
The age of the patient at the first presentation of a soft tissue tumor could be suggestive of a given type of neoplasm. Rhabdomyosarcoma is the most common soft tissue tumor of childhood and accounts for approximately one half of all soft tissue sarcomas in this age group. Approximately 65% of cases occur in children less than 6 years of age. It is less freq...
Table of contents
- Cover
- Contents
- Title
- Copyright
- Foreword
- Preface
- Acknowledgments
- Contributors
- Chapter 1: Clinical Approach in Soft Tissue Tumors
- Chapter 2: Radiological Diagnostic Approach in Soft Tissue Tumors
- Chapter 3: Sampling Procedure, Fine Needle Aspiration (FNA), and Core Needle Biopsy (CNB)
- Chapter 4: Ancillary Techniques
- Chapter 5: Principal Aspects in Fine Needle Aspiration and Core Needle Biopsies
- Chapter 6: Particular Aspects
- Index
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Soft Tissue Tumors by Jerzy Klijanienko,Real Lagace in PDF and/or ePUB format, as well as other popular books in Medicina & Patologia. We have over 1.5 million books available in our catalogue for you to explore.