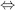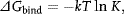Innovative and forward-looking, this volume focuses on recent achievements in this rapidly progressing field and looks at future potential for development. The first part provides a basic understanding of the factors governing protein-ligand interactions, followed by a comparison of key experimental methods (calorimetry, surface plasmon resonance, NMR) used in generating interaction data. The second half of the book is devoted to insilico methods of modeling and predicting molecular recognition and binding, ranging from first principles-based to approximate ones. Here, as elsewhere in the book, emphasis is placed on novel approaches and recent improvements to established methods. The final part looks at unresolved challenges, and the strategies to address them. With the content relevant for all drug classes and therapeutic fields, this is an inspiring and often-consulted guide to the complexity of protein-ligand interaction modeling and analysis for both novices and experts.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Statistical Thermodynamics of Binding and Molecular Recognition Models
Kim A. Sharp
1.1 Introductory Remarks
Equilibrium binding or association of two molecules to form a bimolecular complex, A + B
AB, is a thermodynamic event. This chapter will cover some of the fundamental thermodynamics and statistical mechanics aspects of this event. The aim is to introduce general principles and broad theoretical approaches to the calculation of binding constants, while later chapters will provide examples. Only the noncovalent, bimolecular association under ambient pressure conditions will be considered. However, extension to higher order association involves no additional principles, and extension to high pressure by inclusion of the appropriate pressure–volume work term is straightforward. In terms of the binding reaction above, the association and dissociation constants are defined as K = [AB]/[A][B] and KD = [A][B]/[AB] respectively, where [] indicates concentration. Either K or KD is the primary experimental observable measured in binding reactions. KD is sometimes obtained indirectly by inhibition of binding of a different ligand as a Ki. From a thermodynamic perspective, the information content from K, KD, and Ki is the same.
1.2 The Binding Constant and Free Energy
To connect the experimental observable K to thermodynamics, one often finds in the literature the relationship
(1.1)
where k is the Boltzmann constant, T is the absolute temperature, and ΔGbind is the “absolute” or “standard” binding free energy. Several comments are given to avoid misuse of this expression. First, one cannot properly take the logarithm of a quantity with units such as K, so Eq. (1.1) is implicitly
(1.2)
where Vref is the reference volume in units consistent with the units of concentration in K, that is, 1 l/mol or about 1660 Å3/molecule for molarity units. The choice of Vref is often referred to as the “standard state” problem. Equivalently, one says that ΔGbind is the free energy change when reactants A and B and the product AB are all at the reference concentration. Second, although the units of concentration used in K are almost always moles/liter, this is entirely a convention, so the actual numerical value for ΔGbind obtained from Eq. (1.2) is arbitrary. Put another way, any method for calculating the free energy of binding must explicitly account for a particular choice of Vref before it can meaningfully be compared with experimental values of ΔGbind obtained using Eq. (1.2). Furthermore, ligand efficiency-type measures, such as ΔGbind/n where n is the number of heavy atoms in a ligand or the molecular weight of a ligand [1], can change radically with (arbitrary) choice of concentration units. Of course, differences in ΔGbind can be sensibly compared provided the same reference state concentration is used. Finally, in Eq. (1.2), the free energy actually depends on the ratio of activities of reactants and products, not on concentrations. For neutral ligands and molecules of low charge density at less than micromolar concentrations, the activity and concentration are nearly equal and little error is introduced. However, this is not true for high charge density molecules such as nucleic acids and many of the ligands and proteins that bind to nucleic acids. Here, the activity coefficient can be substantially different from unity even at infinitely low concentration. Indeed, much of the salt dependence of ligand–DNA binding can be treated as an activity coefficient effect [2–4]. The issue of standard state concentrations, the formal relationship between the binding constant and the free energy, and the effect of activity coefficients are all treatable by a consistent statistical mechanical treatment of binding, as described in Section 1.3.
1.3 A Statistical Mechanical Treatment of Binding
Derivation of a general expression for the binding constant follows closely the approach of Luo and Sharp [5], although somewhat different treatments using chemical potentials, which provide the same final result, are given elsewhere [6–8]. It is a statistical mechanical principle that any equilibrium observable can be obtained as an ensemble, or Boltzmann weighted average, of the appropriate quantity. Here, the binding constant K = [AB]/[A][B] is the required observable. Consider a single molecule each of A and B in some volume V (Figure 1.1) and for convenience define a coordinate system centered on B (the target) in a fixed orientation. Over time, the ligand (A) will explore different positions and orientations (poses) relative to B, where r and Ω represent the three position and three orientation coordinates of A with respect to B. Now A and B interact with each other with an energy that depends not only on their relative position (r, Ω) but in general also on the conformations of A, B, and the surrounding solvent. If na, nb, ns are the number of atoms in A, B, and solvent, then the energy is a function of 3na + 3nb + 3ns − 6 coordinates. In principle, one could keep all these degrees of freedom explicit. From a practical standpoint, this would be a complicated and expensive function to evaluate. However, one may integrate over the solvent coordinates and the (3na − 6) +(3nb − 6) internal coordinates so that the interaction between A and B for a given (r, Ω) is described by an interaction potential of mean force (pmf) ω(r, Ω). If one defines the pmf between A and B at infinite separation in their equilibrium conformations to be 0, then ω(r, Ω) is the thermodynamic work of bringing A and B from far apart to some mutual pose (r, Ω), accounting for both solvent effects and internal degrees of free...
Table of contents
Cover
Series Page
Title Page
Copyright
List of Contributors
Preface
A Personal Foreword
Part I: Binding Thermodynamics
Part II: Learning from Biophysical Experiments
Part III: Modeling Protein–Ligand Interactions
Part IV: Challenges in Molecular Recognition
Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
Both plans are available with monthly, semester, or annual billing cycles.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go. Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Protein-Ligand Interactions by Holger Gohlke, Raimund Mannhold,Hugo Kubinyi,Gerd Folkers in PDF and/or ePUB format, as well as other popular books in Medicine & Pharmacology. We have over 1.5 million books available in our catalogue for you to explore.