![]()
PART 1
General Aspects of Lysosomal Storage Diseases
![]()
CHAPTER 1
The Lysosomal System: Physiology and Pathology
Matthew C. Micsenyi and Steven U. Walkley
The Dominick P. Purpura Department of Neuroscience, Albert Einstein College of Medicine, Bronx, NY, USA
Introduction
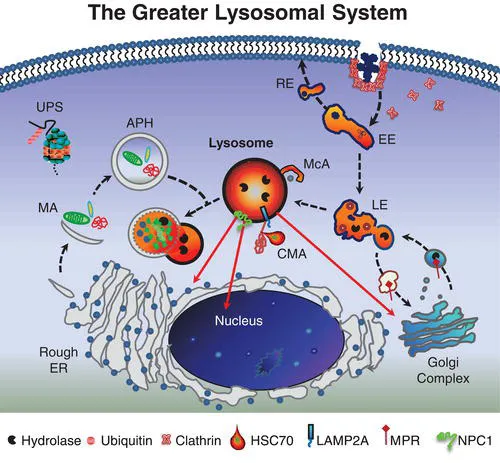
The lysosome and its constituent parts – what has been referred to as the greater lysosomal system [1] – constitute a major metabolic regulatory network in eukaryotic cells. This system includes secretory streams transporting newly synthesized enzymes and other proteins to lysosomes, endosomal and retrosomal streams contributing to signal transduction and related processing, autophagic streams delivering intracellular material for lysosomal degradation, and salvage streams facilitating egress of lysosomal degradation products to other sites in the cell for reutilization. Operating in close parallel are additional proteolytic mechanisms such as the ubiquitin–proteasome system (UPS), which assists in efficient protein turnover. The central coordinator of this remarkable intracellular network is ultimately the lysosome itself, an acidic membrane-bound organelle that functions to degrade and reprocess a vast array of cellular material. Hydrolytic enzymes localized to the lysosomal lumen are optimally active at an acidic pH and have the capacity to degrade most macromolecules including proteins, carbohydrates, lipids, RNA and DNA. Following the breakdown of this material, the resultant amino acids, sugars, simple glycolipids, cholesterol and nucleotides are salvaged by transport through the lysosomal membrane with the aid of specific transporter proteins for delivery to other cell organelles and membranes for subsequent use in biosynthetic processes. Although traditionally depicted as a terminal compartment, this role in recycling molecular precursors brings the importance of the lysosome full circle. Taken as a whole, the greater lysosomal system therefore functions at the very hub of cellular metabolic homeostasis. With the recent discovery of an overarching gene regulatory network referred to as CLEAR (Coordinated Lysosomal Expression and Regulation) and its master gene transcription factor EB (TFEB), many components of the greater lysosomal system have been shown to be linked at the transcriptional level [2]. Indeed, these studies further establish the lysosomal system as a highly efficient and coordinated network. As such, proper lysosomal function is essential since failure of this system leads, inexorably, to catastrophic consequences for cells, organs, and individuals, with nearly 60 different types of lysosomal diseases documented to date (see Classification in Chapter 5).
The greater lysosomal system
Our understanding of the lysosome and its role in cells has evolved significantly since its discovery by Christian de Duve more than 50 years ago. This organelle and the constituent streams or pathways to which it is linked comprise a processing and recycling centre essential to all cells. While each component is typically defined separately, it is important to conceptualize the various parts functioning as a highly orchestrated cellular mechanism (Figure 1.1).
Endocytosis
The endolysosomal pathway consists of the major delivery streams and molecular machinery necessary for the internalization of cell surface and extracellular material linking cells with their external environment. The full scope of the complexity of the endocytic system continues to evolve with the characterization of diverse forms of endocytosis and the elucidation of key molecular components associated with these pathways. Endocytic processes are often grouped by the morphological characteristics of the invagination of the membrane. Clathrin-mediated endocytosis (CME) is defined by clathrin-coated pits localized to the plasmalemma that internalize receptor/cargo complexes into vesicles that are sorted and targeted to various intracellular locations. In the CNS, neurons rely on CME for the cycling of neurotransmitter receptors regulating signaling and activity-dependent neuroplasticity. Clathrin-independent endocytosis has also been described and most often occurs at flask-like invaginations along the plasmalemma called caveolae. Caveolae are long-lived plasma membrane microdomains composed of caveolins, cholesterol, sphingolipids including glycosphingolipids (GSLs) and sphingomyelin, GPI-anchored proteins and various receptor proteins. Such specialized membrane structures which are ultimately processed by the endolysosomal system are known to play a critical role as platforms for cell signalling and as regulators of lipid components within the plasmalemma.
The canonical endocytic pathway progresses along an increasing lumen-acidic gradient from early endosomes retrogradely trafficked from the plasma membrane, to multivesicular bodies or late endosomes, and finally to perinuclear-localized lysosomes. Deviating from this pathway, early endosomes can be recruited back to the plasmalemma or to other organelles as sorting/recycling endosomes. These divergent streams allow for the recycling and reinsertion of cell surface receptors, delivery of signaling ligands throughout the cell, and internalization of membrane components to be reorganized. Such carefully orchestrated processing and its related signal transduction events may be interrupted in diseases of the lysosomal system in which endocytosed components including cholesterol and GSLs accumulate. While this accumulation is typically associated with late endosomes and lysosomes, recent evidence has emerged suggesting additional involvement of early endosomal compartments [3], raising the likelihood that the consequences of lysosomal disease extend well beyond the lysosome itself.
Autophagy
In addition to endocytic pathways, autophagic streams feed into the lysosome and are involved in targeting intracellular material including effete organelles, long-lived proteins and pathogens for degradation [4]. Autophagy, which is often activated following starvation stress, is divided into three distinct subtypes – microautophagy, chaperone-mediated autophagy (CMA) and macroautophagy – defined by the delivery method of substrate to lysosomes. Microautophagy involves the direct engulfment of cytosolic material by the lysosome either by direct invagination of the lysosomal membrane, or projected arm-like extensions that sequester material into intralysosomal vesicles. CMA is selective for soluble monomeric proteins containing the peptide signalling motif Lys-Phe-Glu-Arg-Gln (KFERQ). This motif allows for binding by the heat shock cognate 70 protein (HSC70). HSC70 and co-chaperones then promote protein unfolding and translocation across the lysosomal membrane via the lysosome-associated membrane protein type 2A (LAMP2A). Macroautophagy has been traditionally characterized as a vesicle-mediated bulk degradation mechanism activated in response to nutrient deprivation. Recently, however, it has been shown that macroautophagy is also constitutively active and selective. When macroautophagy is stimulated, double membrane vesicles known as autophagosomes form to engulf cytosolic material. This material, including dysfunctional organelles and oligomerized proteins, is selectively recognized by chaperone complexes and adapter molecules including heat-shock proteins (HSPs), ubiquitin, and p62/SQSTM1 (p62) which allow for the specific uptake of these substrates within autophagosomes. To date several lysosomal diseases have been found to exhibit alterations in autophagy, however the downstream consequences for disease progression remain unclear. Of particular interest is evaluating how impaired autophagy contributes to neurodegenerative processes.
Salvage
Degradation products resulting from lysosomal processing must be efficiently removed from the organelle for utilization elsewhere in the cell. This salvage process involves numerous lysosomal membrane proteins that act as transporters, including cystinosin, sialin, cobalamin transporter and NPC1 protein (see Chapter 17). For example, a defect in the salvage of cobalamin (also known as vitamin B12) leads to cobalamin F-type disease, a disorder characterized in children by megaloblastic anaemia, failure to thrive and neurological deficits [5]. The inabil...