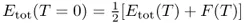While its results normally complement the information obtained by chemical experiments, computer computations can in some cases predict unobserved chemical phenomena Electronic-Structure Computational Methods for Large Systems gives readers a simple description of modern electronic-structure techniques. It shows what techniques are pertinent for particular problems in biotechnology and nanotechnology and provides a balanced treatment of topics that teach strengths and weaknesses, appropriate and inappropriate methods. It's a book that will enhance the your calculating confidence and improve your ability to predict new effects and solve new problems.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
School of Physics and Advanced Materials, University of Technology, Sydney, NSW, Australia
SIESTA provides access to the usual set of properties common to most DFT implementations:
Total energy, charge densities, and potentials
Atomic forces and unit cell stresses
Geometry specification in Cartesian and/or internal z-matrix coordinates
Geometry optimization using the conjugate gradient, modified Broyden and Fire algorithms, and simulated annealing
Total and partial densities of states
Band dispersions
Constant energy, temperature, or pressure molecular dynamics
Simulation of scanning tunneling microscope images according to the Tersoff–Hamann approximation
Electron transport properties using the nonequilibrium Green's function approach
Optical properties and the frequency-dependent dielectric function within the random phase approximation and using first-order time-dependent perturbation theory
Phonon spectrum and vibrational frequencies
Mulliken population analysis
Born charges
In this chapter a number of these properties are discussed through examples relevant to nanoscience and technology. The SIESTA methodology is described in detail in Chapter 2; the present chapter is intended as an accompaniment. The first three examples illustrate the general capabilities of the SIESTA code for problems containing relatively small numbers of atoms and that are amenable to standard diagonalization to solve the self-consistent problem. The last example illustrates the divide-and-conquer linear-scaling capabilities to tackle problems containing large numbers of atoms.
11.1 Ethynylbenzene Adsorption on Au(111)
There has been considerable interest for some time in self-assembled monolayers (SAMs) in nanotechnology. They are relatively easy to prepare on a variety of surfaces, gold being the most common, with a wide range of molecules forming ordered molecular layers.(1–3) They are a useful platform for controlling surface properties and providing functionality with applications in, for example, molecular electronics.(4, 5)
The alkynyl group as method of anchoring SAMs to gold surfaces is a promising candidate to study. It should provide an unbroken conjugated pathway to the gold surface, unlike thiol linkers, and a wide range of terminal alkynes can be synthesized.(6) Ethynylbenzene is a simple representative example of this class of molecule; there is some experimental evidence that it binds to gold surfaces and nanoparticles, although these studies are inconclusive about the nature of the bond.(7, 8) The calculations described below attempt to answer the question of whether this molecule is likely to form SAMs and the likely adsorption geometries and energetics.(9, 10)
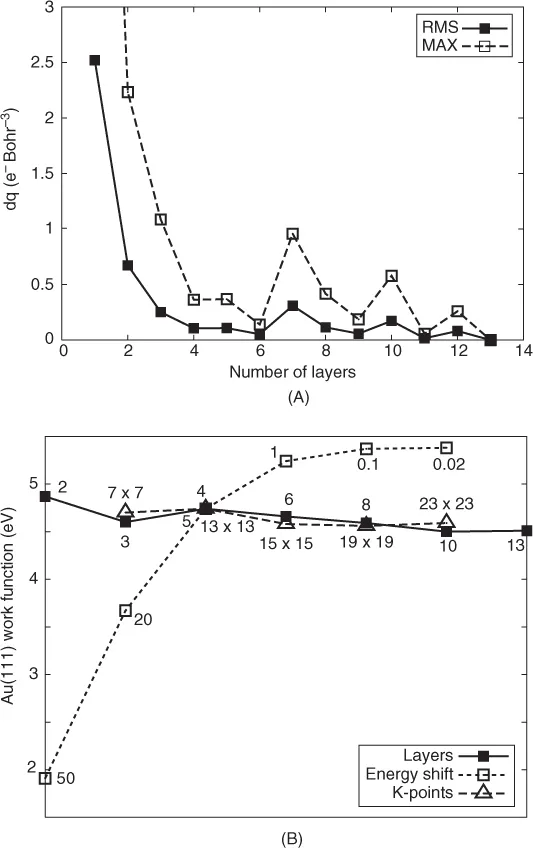
The computational conditions first have to be established and an appropriate representation of the semi-infinite surface in terms of a multilayer slab needs to be determined. The slab needs to contain sufficient layers that the center of the slab is relatively bulklike, or in this particular case so that a molecule adsorbed on one side of the slab is not influenced by the other surface. Conversely, the slab should not be too big, such that the calculations are prohibitively large. Figure 11.1 shows the convergence of surface charge density above the slab layer and convergence of the workfunction for an Au(111) slab as a function of the number of layers. Convergence of the workfunction with two computational parameters, reciprocal space grid (k-grid), and orbital confinement (energy shift) are also shown in Fig. 11.1A. The workfunction is calculated as the difference between the electrostatic potential in vacuum (i.e., at a position in the unit cell far above the surface) and the Fermi level.
The charge density and density difference are extracted from the density matrix (saved to file at each SCF step) using the DENCHAR utility at the points of a user-specified plane, or volume. Charge densities can then be visualized using standard plotting packages. Alternatively, the charge densities and potentials evaluated over the real space grid used to represent the density matrix can be written to file directly from SIESTA by setting the appropriate input flags. These are written unformatted and need to be processed for plotting. The GRID2CUBE utility will generate formatted output from these files in the format of a GAUSSIAN cube file.
The calculations in Fig. 11.1 are for a 1 × 1 unit cell in the plane of the surface, that is, one atom per layer. The equivalent of a double-zeta plus polarization (DZP) basis set is used. A generalized-gradient approximation to the exchange-correlation functional according to Perdew–Burke–Ernzerhof (GGA-PBE)(11) and a real-space integration grid with a 300-Ry cutoff are employed (1 Ry = 0.5 atomic unit of energy = ca.13.6 eV). It is often advisable to use a fine real-space grid to avoid numerical errors; the time penalty for such a grid is not generally a limiting factor. A cutoff of 300 Ry is well converged. A Troullier–Martins pseudopotential(12) with scalar relativistic corrections is used to represent the core Au electrons, with a valence of 5d106s. Cutoff radii for each of the angular momentum channels of the pseudopotential are 2.32 Å for s and p, 1.48 Å for d and f. The quality of these pseudopotentials has been checked in the usual way by comparing against all electron calculations for the atom; they reproduce well the bulk properties of gold (lattice parameter, cohesive energy, and bulk modulus).(13) It is interesting to note that values for the total and cohesive energies of bulk gold do not vary much between a single-zeta plus polarization (SZP) and a DZP basis set, while DZ is considerably worse. Where computational cost is a limiting factor, an SZP basis may be acceptable, although for adsorption energies DZP is probably necessary.
The Au(111) surface is unusual in that it reconstructs to form a
structure with a period of about 63 Å,(14) although there is evidence that this reconstruction is lifted in the presence of adsorbed molecules.(14, 15) More recently, experimental measurements and calculations suggest that thiolate adsorption drives an alternative gold adatom structure and that these adatoms are an integral part of the adsorption motif.(16–18) A detailed analysis of these points is beyond the scope of the present chapter, where we are more interested in demonstrating the utility of the SIESTA methodology. Accordingly, a bulk terminated Au(111) surface is assumed.
Temperature smearing of the electron occupation is employed in these calculations to assist convergence of the SCF steps. Both the standard Fermi–Dirac function and the function proposed by Methfessel and Paxton(19) are implemented in SIESTA. In this case it is the free energy F(T) that is minimized during self-consistency. The total energy in the athermal limit is then approximated by the expression
11.1
The degree of smearing is determined by specifying a fictitious temperature to the electron distribution; in this case, a temperature corresponding to 25 meV is used.
Charge density close to the slab surface has converged by four layers and thereafter oscillates slightly. The charge density should be a reasonable indicator of how the adsorption properties will converge. The workfunction is less sensitive to the number of slab layers and the k-grid. Again four layers and a 15 × 15 k-grid are reasonably converged. Only one k-point is required perpendicular to the surface because there is no periodicity in this direction. The workfunction is very sensitive to the energy shift, with values as small as 0.1 mRy required for good convergence. This level is impractical for realistic surface adsorption calculations, as it is extremely time intensive. It is worth noting that the converged value of the workfunction calculated here is 5.13 eV, compared with an experimental value of 5.31 eV.(20)
The conclus...
Table of contents
Cover
Title Page
Copyright
Dedication Page
Preface
Contributors
Part A: DFT: The Basic Workhorse
Part B: Higher-Accuracy Methods
Part C: More-Economical Methods
Part D: Advanced Applications
Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
Both plans are available with monthly, semester, or annual billing cycles.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go. Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Computational Methods for Large Systems by Jeffrey R. Reimers in PDF and/or ePUB format, as well as other popular books in Ciencias físicas & Química física y teórica. We have over 1.5 million books available in our catalogue for you to explore.