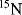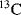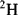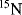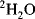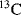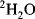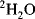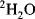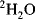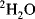Nuclear Magnetic Resonance (NMR) spectroscopy, a physical phenomenon based upon the magnetic properties of certain atomic nuclei, has found a wide range of applications in life sciences over recent decades. This up-to-date volume covers NMR techniques and their application to proteins, with a focus on practical details. Providing newcomers to NMR with practical guidance to carry out successful experiments with proteins and analyze the resulting spectra, those familiar with the chemical applications of NMR will also find it useful in understanding the special requirements of protein NMR.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Sample Preparation, Data Collection and Processing
Frederick W. Muskett
Purgamentum init, exit purgamentum
1.1 Introduction
The power of NMR spectroscopy for the analysis of biological macromolecules is undisputed. During the last two decades, the development of spectrometers and the experiments they perform, software, and the molecular biological techniques for the expression and purification of proteins have progressed at a formidable rate. Enrichment of molecules in the three major isotopes used in NMR (
,
and
) is now commonplace and the cost is no longer prohibitive. The software used to analyse the plethora of data we can generate makes spectral assignment and the extraction of data straightforward for all but the most challenging systems. With all these developments it is easy to forget some of the more fundamental requirements for obtaining good quality NMR data, namely a good sample and a well-set-up NMR experiment.
1.2 Sample Preparation
The first, and possibly one of the most important, steps before embarking on an NMR-based project is the preparation of the sample. Spending some time optimising sample conditions for concentration, ionic strength, pH and temperature before collecting large amounts of data will pay dividends, particularly if the sample is difficult and expensive to produce. Ideally, the optimised sample will not only give the best possible NMR data, but will also have long-term stability as, assuming the project requires backbone and side-chain assignments, the total acquisition time required can be in the order of several weeks.
The following sections will outline the general requirements of a biological sample that is to be used to record NMR data. The assumption has been made that full resonance assignment is required; however, these guidelines could, and probably should, be applied to all samples regardless of the intention of the experiments.
1.2.1 Initial Considerations
This optimisation can be performed in the NMR spectrometer but much can be done using other biophysical techniques such as circular dichroism or fluorescence spectroscopy. These methods require much lower concentrations and do not require isotopic enrichment. The effects of buffer composition on secondary structure content and the melting temperature of the sample give a useful starting point. Once the initial conditions have been determined, final optimisation in the spectrometer can begin. If the sample is a protein, although much can be learned from a simple one-dimensional proton experiment, by far the most useful experiment is the
-edited HSQC. This type of experiment removes a great deal of resonance overlap, allowing the user to see in much more detail the effects of varying pH, ionic strength and temperature.
NMR has intrinsically poor sensitivity and, as a result, the concentration of the sample needs to be in the millimolar range. For a conventional room temperature probe ideally the sample concentration needs to be ≥1 mM but can be as low as 0.5 mM. With the development of cryogenically cooled probes this concentration can be reduced to ≥0.2 mM and given the right sample can be as low as 0.05 mM (depending on the experiments performed). Whilst the sample can be exchanged into the buffer intended for NMR experiments in the last step of purification (usually gel filtration chromatography) it is rarely at the concentration required. The two main methods used to increase concentration are lyophilisation with subsequent re-suspension in a lower volume and ultra-filtration, or a combination of the two. Unfortunately, whether a sample will survive either method cannot be predicted; in the end one must simply try and see what happens. However, lyophilisation is generally considered the more dangerous of the two. The number and type of disposable ultra-filtration devices on the market is large, each with their own characteristics regarding compatibility with a particular sample and the effective volumes with which they can be used; again, try and see what happens. As a final step, either passing the sample though a 0.2 μm filter or centrifuging in a benchtop micro-centrifuge, to remove any insoluble material or dust, will greatly help sample homogeneity.
With modern solvent suppression techniques that can effectively eliminate the
110 M protons from the water signal, dissolving the sample in
would, at first, no longer seem to be required. However, such samples are still important in recording the experiments designed to allow assignment of protein side-chain resonances and for
-edited nuclear Overhauser effect (NOE) experiments. Even the most efficient solvent suppression techniques still leave residual solvent signal and at the same time suppress or distort the signals of interest in that area of the spectrum. In addition, the use of
allows one to carry out experiments in which the coherences are recorded in the directly detected dimension where they have the highest resolution. These two advantages alone outweigh the effort required to transfer the sample into
. Methods for exchanging the solvent for
are the same as for concentrating samples, either lyophilisation or repeated concentration and dilution with
. Alternatively, if the sample is unlikely to survive those methods, the sample can be passed down a short de-salting gel-filtration column that has been pre-equilibrated in
.
1.2.2 Additives
As many NMR experiments require hours or days to complete, addition of anti-microbial agents is highly recommended. Sodium azide at a concentration of 0.02 % w/v is an almost universal method; however, in the rare cases where the azide ion interacts with the sample (e.g. some cytochromes) micromolar concentrations of an antibiotic such as ampicillin or chloramphenicol can be substituted. EDTA or AEBSF™ are frequently added to NMR samples at a concentration of 0.1–5 mM in order to reduce proteolysis. However, these compounds have nonexchangeable protons that can interfere with the spectrum of the sample. Excessive use of these compounds is best avoided, a better approach being to improve the purification protocol. If the protein sample contains free cysteines reducing agents such as DTT or TCEP™ are required to stop the protein forming dimers or multimers, which can result in precipitation. In addition, degassing the sample can help; however samples inevitably re-dissolve oxygen during subsequent sample manipulations or during the course of the NMR experiment unless special care is taken to seal the NMR tubes.
1.2.3 Sample Conditions
Although the primary choice of buffer must be that which promotes long-term stability of the sample, some buffer salts are more convenient for NMR than others. As buffer concentrations are typically between 10–50 mM, any covalently bonded protons in the buffer will give rise to sharp and obtrusive signals in the spectrum. This has resulted in phosphate buffer being the primary choice if its buffering range is appropriate to your sample and if it does not interact with your protein – though many proteins bind ligands containing phosphate groups, from ATP to phosphoproteins and DNA, and may bind inorganic phosphate weakly. Otherwise, many of the more common buffer salts are available with deuterium replacing the nonlabile protons. When selecting a pH it should be borne in mind that the exchange rates of amide protons are such that pH values between 3 and 7 are most conducive to observing the signals arising from these groups.
For many biological samples, the addition of salts (typically sodium chloride) to the buffer increases solubility and decreases aggregation. Unfortunately, in NMR the dielectric losses at high ionic strength (greater than 150 mM) are severe, particularly in cryogenically cooled probes and at high magnetic fields. Such losses can be dramatic, as degrading the signal-to-noise ratio twofold results in a fourfold increase in acquisition time to achieve comparable spectra. Recently, alternatives to traditional buffer systems have be proposed, such as dipolar ions [1], low conductivity buffers [2] or the use of ‘solubilising salts’ [3]. The general applicability of these alternatives is yet to be realised; however, they should be investigated if the sample has low solubility and/or requires high ionic strength for stability.
The final parameter to consider in optimising the sample conditions is the temperature at which the experiments are to be performed. As the temperature increases, the correlation time of the molecules decreases and so the resonances become narrower. In addition, varying the temperature will lead to changes in the chemical shift of temperature sensitive groups and may help to resolve any resonance overlaps. Typically, NMR experiments are performed in the temperature range of 293–308 K but if you have determined the thermal stability of the sample in advance (i.e. by CD spectroscopy), you will have a better idea of the attainable upper limit.
1.2.4 Special Cases
There are two types of sample that require extra attention: integral membrane proteins and samples intended for ligand titrations.
The use of solution NMR methods to study membrane proteins, although not mainstream, is now feasible. The solubilisation of integral membrane proteins in detergent micelles is relatively straightforward and the procedures for preparing membrane protein samples are essentially as described. There is much debate about which detergents are best for preparing NMR samples and it is apparent that no one detergent will suit all proteins. As a result, screening of several different detergent types at different concentrations is required. In addition, significant improvement of the spectrum can be achieved by using sample temperatures significantly higher than those used for soluble proteins (>310 K). The reader is referred to some excellent reviews on sample optimisation [4–6].
The study of ligand interactions via NMR is a well-established technique, enabling the identification of a specific binding site and the determination of kinetic information (see Chapter 7). The usual method for obtaining this information is to run successive spectra with increasing concentrations of ligand whilst observing the spectral changes that result. However, there is a danger that addition of the ligand will lead to changes in the sample conditions other than those due to ligand binding, resulting in artefactual/artificial changes in the spectrum. It can be difficult to obtain identical buffer conditions on mixing different proportions of two or more samples even if they have been dialysed against the same buffer but in separate dialysis tubes, as many biological macromolecules have a high affinity for electrolytes. In addition, if the ligand is a small molecule it may be difficult to solubilise and impossible to dialyse into the same buffer as the macromolecule.
The major concern in mixing two samples together or adding a ligand to a sample is a change in pH, as this alone can result in considerable chemical shift changes in the molecule of interest, as any ionisable groups change state. Fortunately, this property of ionisable groups can be used to monitor the pH of the NMR sample. Addition of a small molecule (e.g. imidazole) to the sample can be used to monitor the sample's pH as additions of ligand are made. Any pH shift will become immediately apparent as the chemical shift of the imidazole resonances will change. A number of these molecules have recently been characterised and the reader is referred to this article for more details [7].
In...
Table of contents
Cover
Title Page
Copyright
List of Contributors
Introduction
Chapter 1: Sample Preparation, Data Collection and Processing
Chapter 2: Isotope Labelling
Chapter 3: Resonance Assignments
Chapter 4: Measurement of Structural Restraints
Chapter 5: Calculation of Structures from NMR Restraints
Chapter 6: Paramagnetic Tools in Protein NMR
Chapter 7: Structural and Dynamic Information on Ligand Binding
Chapter 8: Macromolecular Complexes
Chapter 9: Studying Partially Folded and Intrinsically Disordered Proteins Using NMR Residual Dipolar Couplings
Color Plates
Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
Both plans are available with monthly, semester, or annual billing cycles.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go. Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Protein NMR Spectroscopy by Lu-Yun Lian, Gordon Roberts, Lu-Yun Lian,Gordon Roberts, Gordon Roberts, Lu-Yun Lian in PDF and/or ePUB format, as well as other popular books in Physical Sciences & Spectroscopy & Spectrum Analysis. We have over 1.5 million books available in our catalogue for you to explore.