
- 203 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
The Protein Folding Problem
About this book
Proteins in living systems carry out a great variety of specific functions, each of which depends on the precise three-dimensional structure of a particular protein. Proteins are synthesized in the form of a flexible polypeptide chain that is capable of assuming a vast number of configurations; the transformation of this chain into a specific, relatively rigid three-dimensional structure is called folding--a remarkable process of self-organization. It is known that the amino acid sequences of some proteins have sufficient information to determine their three-dimensional structures. There are other proteins whose folding requires additional information beyond that found in the sequence of the mature protein. This book introduces the central problem of folding mechanisms as well as a number of other closely related issues. This book is neither a textbook nor a treatise. Rather, it is an attempt by several investigators to convey the excitement and challenges of those aspects of the folding problem in which they are actively engaged. The contributors give brief introductions to protein folding from the perspectives of molecular architecture, stability and dynamics, phage genetics, DNA exons, general physiology, and natural selection. They point out emerging new directions, including the suggestion of a class of diseases that result from protein folding defects.
Trusted by 375,005 students
Access to over 1 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
1.
What Do the Folds in Proteins Look Like?
Proteins are at the interface between chemistry and biology: on the one hand they are among the largest of chemically well-defined molecules, while on the other hand they are perhaps the smallest systems which display the complexity, quirkiness, and downright intractability of living systems. One reason for the fascination of the protein folding problem is that it represents an unusually concrete and limited case of the whole problem of reductionism. An unfolded protein is clearly a chemical object: a backbone of exactly repeated simple units each with one of an alphabet of 20 possible side chains. Its properties are relatively dull and are quite predictable by summing up the properties of its components. A folded protein, on the other hand, in addition to complexity and unpredictability, has acquired meaning: unity, controlled interaction with other systems, and biologically significant function. In many cases one can watch a protein undergo the spontaneous transition from randomness to directed functionality in the space of a few minutes. There is apparently no extra information hidden within the starting state, so we feel that understanding the rules of the transformation would teach us worthwhile lessons about hierarchical organization, cooperative properties, and exactly how an organic whole becomes so much more than a sum of its parts.
As an introduction to the general problem of protein folding, I would like to summarize some characteristics of the final folded state of proteins, trying to emphasize properties that seem likely to be related to the pathway by which they arrived there.
It is rather misleading, of course, to speak of a single folded state, because the native protein is a dynamic object that fluctuates around one or a few preferred conformations. The largest of those fluctuations are presumably related to the final steps of folding or the first steps of unfolding. Figure 1 shows all non-hydrogen atoms in the preferred conformation of pancreatic trypsin inhibitor (as determined by X-ray
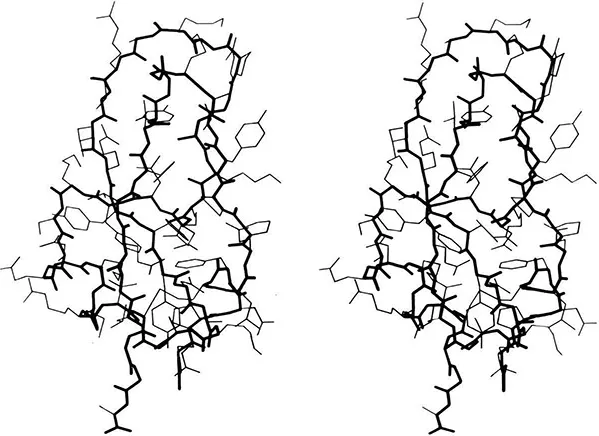
Figure 1. Stereo drawing of all non-hydrogen atoms in the x-ray crystal structure of pancreatic trypsin inhibitor (17). Backbone is in heavy lines and side chains in lighter lines. Coordinates for this and most other figures are from the Protein Data Bank (18).
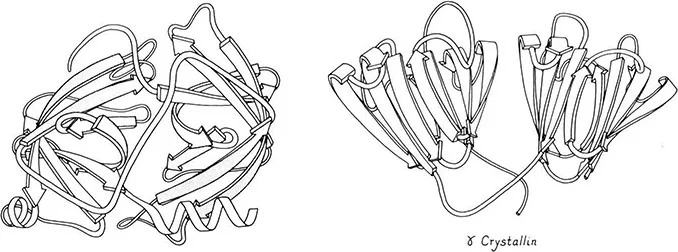
Figure 2. Proteins with two very similar domains. Left: elastase (19), with the domains closely packed into a spherical overall subunit. Right: gamma crystallin (20), with its two domains well separated.
crystallography), to illustrate the complexity and asymmetry of the native conformation. The stick-figure model allows one to look through the molecule and to visualize the covalent connectivity which is a crucial part of folding. However, the internal interactions of a protein with itself that constitute the folding process, and the external interactions with the environment that include its functionality, all occur at the surfaces of the atoms. There is a highly complementary interlocking of atom surfaces inside a protein, with only a little open space left. The final readjustments which allow everything to settle down in the position of best fit is presumably one of the last steps in folding, although once such complementarity is achieved it is not necessarily the first thing to be lost on unfolding.
The most obvious, crucial task that must be accomplished by the protein folding process is arrangement of the backbone into an approximately correct tertiary structure. Although those tertiary structures show a wide variety there is considerable order within that variety, some of which may be the result of restrictions on favorable folding pathways. Within the four major categories there are 9 or 10 rather specific structural patterns, which together account for about 3/4ths of the known protein structures (1). We will survey these common structural patterns, trying to recognize the footprints left there by folding requirements.
Large proteins cure commonly divided into domains: contiguous stretches of the sequence which are also contiguous in 3 dimensions (2). Domains usually resemble other entire single-domain proteins. They sometimes can move as independent rigid bodies. Domains are often assumed (and in a few cases proven) to fold up independently and thereby to make the folding of large proteins fast enough to be biologically useful. The commonest domain size is 100 to 200 residues, but they vary from about 40 to at least 400. Figure 2 shows schematic backbone drawings of elastase and of gamma crystallin, each of which has two very similar domains. Since the domains within one protein are often completely different, however (as shown for papain in Figure 3), classification of tertiary structure is much more straightforward if done one domain at a time. Within each domain we will pay attention to the type, amount, and organization of secondary structure, the number and shape of the major layers of backbone versus hydro-phobic cores, and the topology of connections among the individual chains in a layer. The structures are illustrated by highly simplified schematic drawings of the polypeptide backbone in which alpha-helices are shown as spiral ribbons, beta strands as arrows, non-repetitive loops as smoothed ropes and disulfides as zigzags. All the drawings are to the same scale and within each structural class they are usually shown from an equivalent direction of view.
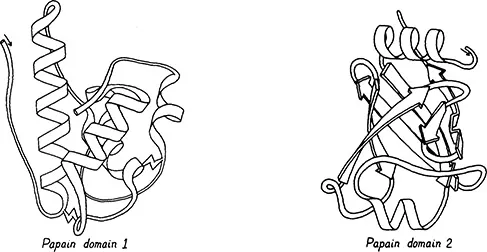
Figure 3. Papain (21), a protein with two entirely different domains. Left: domain 1, which is all helical. Right: domain 2, which is almost entirely a barrel of beta sheet.
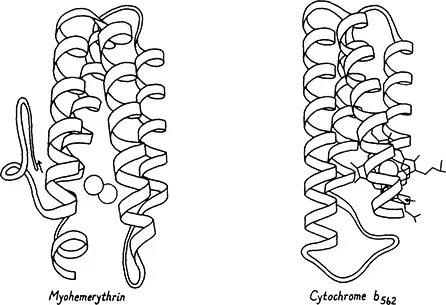
Figure 4. Up-and-down helix bundle structures (antiparallel alpha category): myohemerythrin (22) and cytochrome b562 (23).
The 4 major categories of tertiary structure are:
- Antiparallel alpha
- Parallel alpha/beta
- Antiparallel beta
- Small irregular
It is worth noting, to begin with, that there are no all-alpha (or all-beta) structures in which the elements of each layer are parallel to one another. That would presumably be possible for a 2-layer structure in its final form, but would require the chain to fold by winding first up in one layer and then down in the other, like a ball of string. Apparently that does not happen in such a straightforward form, although later we will find evidence suggesting that more complex elements may fold by winding up as loops.
Antiparallel Alpha Proteins
Within the first major category, the antiparallel alpha, the simplest type of structure is the up-and-down helix bundle: an approximate cylinder of alpha helices with each connected to its nearest neighbor, so that the chain goes up one helix, moves to the next one and down it, over by one and up, etc., all around the cylinder. Many of these structures have only four helices, such as the myohemerythrin and cytochrome b562 shown in Figure 4. Bacteriorhodopsin is a bundle of 7 alpha helices spanning the membrane, and it probably has an up-and-down connectivity (see chapter 5). The prevalence of nearest-neighbor connections between elements of secondary structure, as seen in these up-and-down helix bundles, is one of the most general regularities in protein structure. This has frequently been explained (e.g., 2) by the simple statistical fact that two pieces of backbone can come together more rapidly if they are neighbors in the sequence than if they are widely separated.
The second simplest organization of antiparallel helices has at least one non-nearest-neighbor connection in which the chain goes across either the top or bottom of the cylinder of helices. This does not happen in any of the 4-helix bundles, but it may with 5 or more helices as shown for the hemoglobin beta chain and the second domain of thermolysin in Figure 5. In analogy to the beta barrels described below, this second pattern of antiparallel alpha structure is called a Greek key helix bundle. They are much less common than up-and-down helix bundles, they have no characteristic handedness, and the examples so far each include only one non-nearest-neighbor connection. The distribution of topologies found in helix bundle proteins is consistent with the hypothesis that alpha helices associate as near-neighbor pairs independently of what pairs have already formed.
In each of the major structural categories there is a
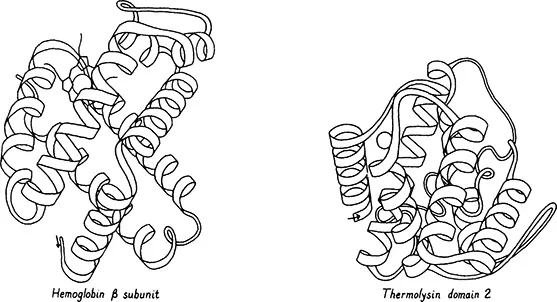
Figure 5. Greek key helix bundle structures (antiparallel alpha category): hemoglobin (24) and thermolysin domain 2 (25).
Figure 6. A miscellaneou...
Table of contents
- Cover
- Half Title
- Series Page
- Title
- Copyright
- Contents
- About the Editor and Authors
- Preface
- 1 What Do the Folds in Proteins Look Like?
- 2 Modular Processes and Natural Selection for Rapid Folding
- 3 Hierarchical Structure and Assembly of Type I Collagen
- 4 Stability and Dynamics of Globular Proteins
- 5 On the Folding and Insertion of Globular Membrane Proteins
- 6 Use of Temperature Sensitive Mutants to Dissect Pathways of Protein Folding and Subunit Interaction
- 7 Exons and Domains in Relation to Protein Folding
- 8 Principles and Problems of Biological Growth
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.4M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access The Protein Folding Problem by Donald B Wetlaufer in PDF and/or ePUB format, as well as other popular books in Social Sciences & Sociology. We have over one million books available in our catalogue for you to explore.