Molecular dynamics simulation is a significant technique to gain insight into the mechanical behavior of nanostructured (NS) materials and associated underlying deformation mechanisms at the atomic scale. The purpose of this book is to detect and correlate critically current achievements and properly assess the state of the art in the mechanical behavior study of NS material in the perspective of the atomic scale simulation of the deformation process. More precisely, the book aims to provide representative examples of mechanical behavior studies carried out using molecular dynamics simulations, which provide contributory research findings toward progress in the field of NS material technology.

eBook - ePub
Molecular Dynamics Simulation of Nanostructured Materials
An Understanding of Mechanical Behavior
- 314 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Molecular Dynamics Simulation of Nanostructured Materials
An Understanding of Mechanical Behavior
About this book
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
1 | Structural Description of Materials |
1.1 ATOMIC ARRANGEMENTS IN MATERIALS
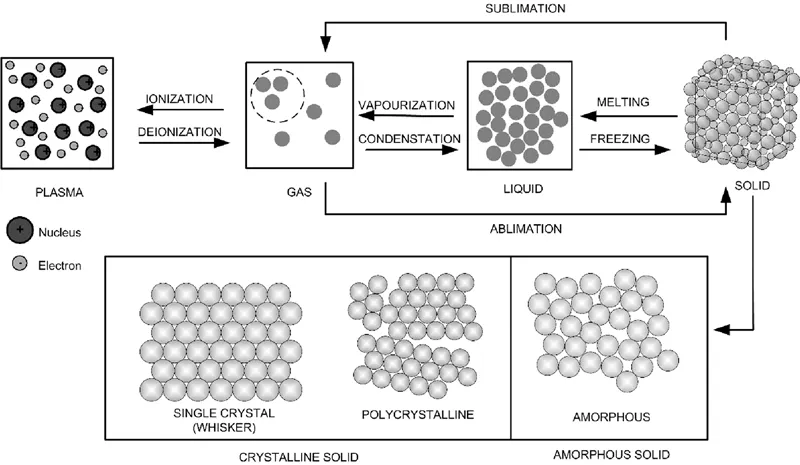
Materials are made up of atoms, molecules, and ions present in different states such as gas, liquid, and solid. According to the kinetic molecular theory, the atoms/molecules in solids vibrate and rotate at their respective places instead of moving. Solids have stronger attractive forces to hold molecules/atoms in contrast to liquids and gases. Solids acquire definite shapes and volumes and can also withstand some amount of shear forces without undergoing any deformation. Furthermore, solids can be broadly classified into two major categories, crystalline solids and amorphous solids, which are shown in Figure 1.1. In crystalline solids, the atoms, ions, or molecules are arranged in a regular, well-defined periodic manner. In amorphous solids, the arrangement of atoms, ions, or molecules is disordered, with lacking periodicity.
Crystalline solids can be divided into following four categories based on the types of particles (atom/ion/molecule) present and the types of bonding between the particles:
1. Ionic solids
2. Metallic solids
3. Covalent network solids
4. Molecular solids
The constituent particles, type of effective binding forces, and properties of different crystalline solids along with examples are presented in Table 1.1.
1.1.1 PERIODICITY IN CRYSTALS AND SYMMETRY ELEMENTS
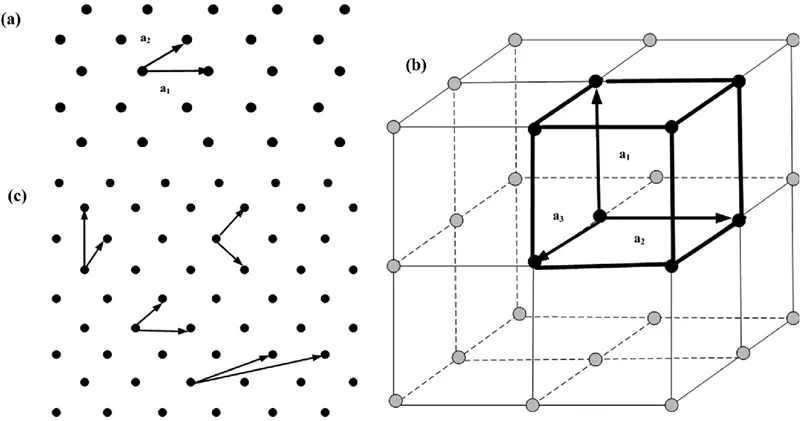
The basic difference between crystals and non-crystals is that atomic arrangement in crystals is in the form of a periodic array in a space. Such kind of periodic array constitutes a three-dimensional (3D) representative unit of a structure, where a group of atoms/molecules is repeated after regular intervals. Such a 3D array of points having identical surroundings may be defined as space lattice. If the periodicity in the structure along a line is a1, then position of any lattice point along the line can be identified by a translation operation, ru = ua1. In case of two-dimensional (2D) and 3D arrangements, similarly, ruv = ua1 + va2 and ruvw = ua1 + va2 + wa3 will be translational vectors, respectively, where u, v, and w are the integers. Figure 1.2a,b,c illustrates 2D and 3D cubic lattices. Unit cell is the smallest unit of volume that permits identical cells to be stacked together in a lattice space; therefore, the entire crystal lattice can be created by repeating the pattern of the unit cell over and over in all directions. If a unit cell is constructed without any overlap with another unit cell and is also free from any kind of defect, then it is called a primitive unit cell. A body is considered to be symmetrical when it is reproduced by certain symmetry operations such as translation, rotation, reflection, and inversion.
Type of Solid | Form of Unit Particles | Forces Between Particles | Solubility | Examples |
|---|---|---|---|---|
Molecular | Atoms or molecules | London dispersion forces, dipole-dipole forces, hydrogen bonds | Depends on the polarity of the molecules | Argon (Ar), methane (CH4), sucrose (C12H22O11), dry ice (CO2) |
Covalent network | Atoms connected in a network of covalent bonds | Covalent bonds | Insoluble in all solvents | Diamond (C) and quartz (SiO2) |
Ionic | Positive and negative ions | Electrostatic attractions | Soluble in polar solvents | Typical salts such as NaCl and Ca(NO3)2 |
Metallic | Atoms | Metallic bonds | Insoluble in all solvents | All metallic elements: Cu, Fe, Al, Pt, etc |
1.1.2 CRYSTAL LATTICES AND STRUCTURES
There are numerous ways in which the atoms can be arranged periodically in different lattices. These are known as the Bravais lattices. The combination of lattice (how to repeat) and motif (what to repeat) constitutes the crystal structure. A perfect crystal is the one in which all its lattice points are occupied by atoms, without any defects. However, such a scenario is not practically possible, as defects exist in the materials. In entirety, there are 7 crystal systems and 14 unique and non-repeating Bravais lattices, which are listed in Table 1.2.
Sl. No. | Crystal System | Relation Between the Sides | Relation Between the Angles | Bravais Lattices, Number and Types |
|---|---|---|---|---|
1. | Cubic | a = b = c | α = β = γ = 90° | 3 = S, BC, FC |
2. | Tetragonal | a = b ≠ c | α = β = γ = 90° | 2 = S, BC |
3. | Hexagonal | a = b ≠ c | α = β = 90°, γ = 120° | 1 = S |
4. | Orthorhombic | a ≠ b ≠ c | α = β = γ = 90° | 4 = S, BC, FC, EC |
5. | Rhombohedral | a = b = c | α = β = γ ≠ 90° | 1 = S |
6. | Monoclinic | a ≠ b ≠ c | α = β = 90°, γ ≠ 120° | 2 = S, EC |
7. | Triclinic | a ≠ b ≠ c | α ≠ β ≠ γ ≠ 90° | 1 = S |
S: simple, BC: body center, FC: face center, EC: end center.
The most common primitive unit cells include Wigner-Seitz cell. The construction of the Wigner-Seitz cell is as follows [1]:
• Draw lines in order to connect a lattice point to all its neighboring points present in the lattice.
• Bisect each constructed line with a plane normal to this line, as presented in Figure 1.3a.
• The Wigner-Seitz cell is the smallest polyhedron bound by these planes. Figure 1.3b illustrates the Wigner-Seitz cell for a body-centered cubic Bravais lattice, where square and hexagonal faces can be seen.
Based on the symmetry considerations, 2D Bravais lattices can be classified into five categories. The most common lowest symmetry lattice includes the oblique lattice, which is invariant when rotated at an angle of π. There are four special lattice types that are invariant under numerous rotations and reflection symmetry operators. The constraints on the unit cell axes and angles for the aforementioned special lattice types are listed in Figure 1.3c, for example, monolayer materials such as graphene and boron. They may be quite large and contain many unit cells (quasi-infinite), or they may be of finite size or long ribbons of a nanoscale width.
Considering symmetry, 3D Bravais lattices can be assorted into 14 types. They are classified into seven types of unit cells based on their restrictions on the unit cell axes and angles for each of the 3D Bravais lattice types. For example, the cubic lattice includes simple cubic, body-centered cubic, and face-centered cubic. Here, the triclinic lattice is the most common, and there are 13 more special lattice types.
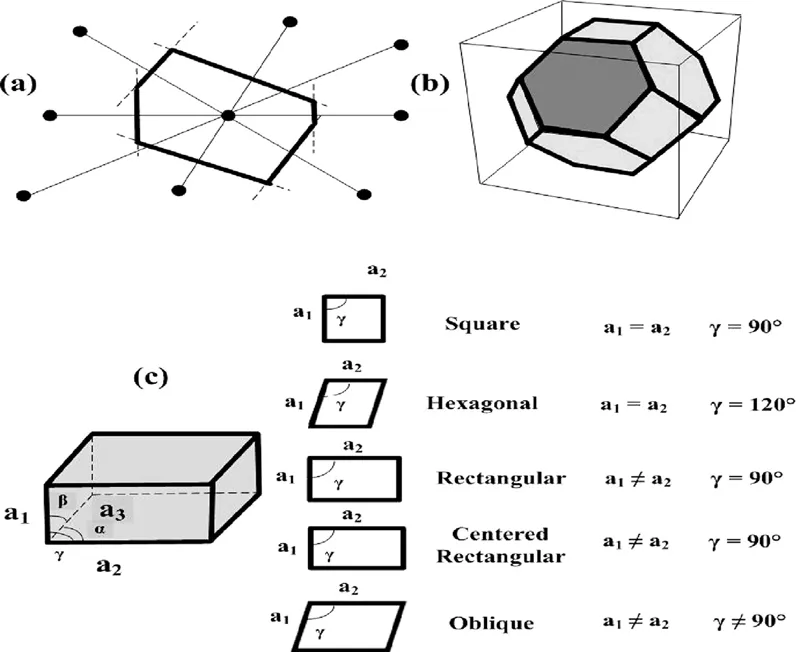
Table of contents
- Cover
- Half Title
- Title Page
- Copyright Page
- Dedication
- Table of Contents
- Preface
- Authors
- Chapter 1 Structural Description of Materials
- Chapter 2 Mechanical Behavior of Materials
- Chapter 3 Creep and Fatigue Behavior of Materials
- Chapter 4 Mechanical Behavior of Nanostructured Materials
- Chapter 5 Basics of Molecular Dynamics Simulation
- Chapter 6 Stress-Strain Behavior Investigation by Molecular Dynamic (MD) Simulation
- Chapter 7 Fracture Simulations Using Molecular Dynamics (MD)
- Chapter 8 Creep Behavior Investigation by Molecular Dynamics (MD) Simulation
- Chapter 9 Fatigue Behavior Investigation by Molecular Dynamics (MD) Simulation
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Molecular Dynamics Simulation of Nanostructured Materials by Snehanshu Pal,Bankim Chandra Ray in PDF and/or ePUB format, as well as other popular books in Technology & Engineering & Mathematics General. We have over 1.5 million books available in our catalogue for you to explore.