
- 292 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
About this book
A thoroughly updated version of the successful first edition with a new chapter on Real-Time PCR, more prokaryotic applications, and more detail in the complex mutagenesis sections. Information on PCR applications in genomics and proteomics have been expanded and integrated throughout the text. There is also advice on available products a
Trusted by 375,005 students
Access to over 1 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Subtopic
BiologyIndex
Biological Sciences1
An introduction to PCR
1.1 Introduction: PCR, a ‘DNA photocopier’
Does it really work? It is so simple! Why did I not think of it? These thoughts were probably typical of most molecular biologists on reading early reports of the polymerase chain reaction or PCR as it is more commonly called. PCR uses a few basic everyday molecular biology reagents to make large numbers of copies of a specific DNA fragment in a test-tube. PCR has been called a ‘DNA photocopier’. While the concept is simple, PCR is a complicated process with many reactants. The concentration of template DNA is initially very low but its concentration increases dramatically as the reaction proceeds and the product molecules become new templates. Other reactants, such as dNTPs and primers, are at concentrations that hardly change during the reaction, while some reactants, such as DNA polymerase, can become limiting. There are significant changes in temperature and pH and therefore dramatic fluctuations in the dynamics of a range of molecular interactions. So, PCR is really a very complex process, but one with tremendous power and versatility for DNA manipulation and analysis.
In the relatively short time since its invention by Kary Mullis, PCR has revolutionized our approach to molecular biology. The impact of PCR on biological and medical research has been like a supercharger in an engine, dramatically speeding the rate of progress of the study of genes and genomes. Using PCR we can now isolate essentially any gene from any organism. It has become a cornerstone of genome sequencing projects, used both for determining DNA sequence data and for the subsequent study of putative genes and their products by high throughput screening methodologies. Having isolated a target gene we can use PCR to tailor its sequence to allow cloning or mutagenesis or we can establish diagnostic tests to detect mutant forms of the gene. PCR has become a routine laboratory technique whose apparent simplicity and ease of use has allowed nonmolecular biology labs to access the power of molecular biology. There are many scientific papers describing new applications or new methods of PCR. Many commercial products and kits have been launched for PCR applications in research and for PCR-based diagnostics and some of these will be discussed in later chapters.
1.2 PCR involves DNA synthesis
PCR copies DNA in the test-tube and uses the basic elements of the natural DNA synthesis and replication processes. In a living cell a highly complex system involving many different proteins is necessary to replicate the complete genome. In simplistic terms, the DNA is unwound and each strand of the parent molecule is used as a template to produce a comple- mentary ‘daughter’ strand. This copying relies on the ability of nucleotides to base pair according to the well-known Watson and Crick rules; A always pairs with T and G always pairs with C. The template strand therefore specifies the base sequence of the new complementary DNA strand. A large number of proteins and other molecules, such as RNA primers, are required to ensure that the process of DNA replication occurs efficiently with high fidelity, which means with few mistakes, and in a tightly regulated manner. DNA synthesis by a DNA polymerase must be ‘primed’, meaning we need to supply a short DNA sequence called a primer that is complementary to a template sequence. Primers are synthetically produced DNA sequences usually around 20 nucleotides long. The DNA polymerase will add nucleotides to the free 3′-OH of this primer according to the normal base pairing rules (Figure 1.1 ).
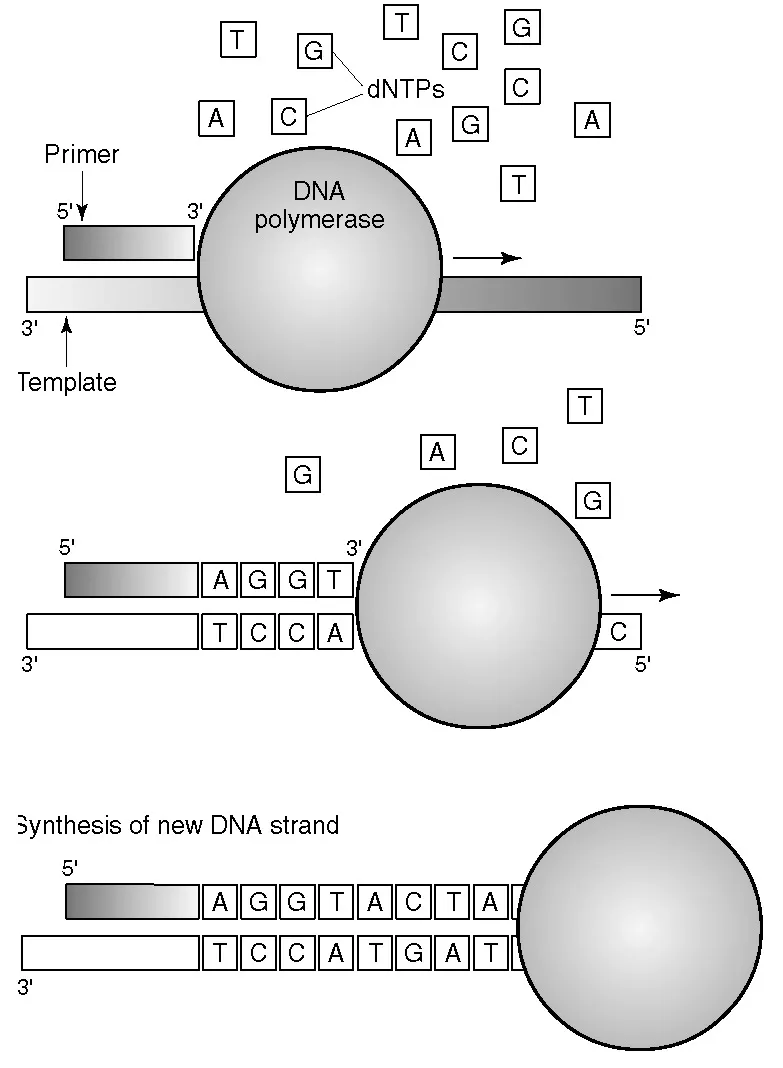
Figure 1.1
Primer extension by a DNA polymerase. The primer anneals to a complementary sequence on the template strand and the DNA polymerase uses the template sequence to extend the primer by incorporation of the correct deoxynucleotide (dNTP) according to base pairing rules.
PCR requires only some of the components of the complex replication machinery to copy short fragments of DNA in a simple buffer system in a test tube. Unwinding of the DNA in the cell uses a multi-component complex involving a variety of enzymes and proteins, but in PCR this is replaced simply by a heating step to break the hydrogen bonds between the base pairs of the DNA duplex, a process called denaturation.
Following template denaturation two sequence-specific oligonucleotide primers bind to their complementary sequences on the template DNA strands according to normal base pairing rules (Figure 1.2). These primers define the region of template to be copied. DNA polymerase then begins to add deoxynucleotides to the 3′-OH group of both primers producing new duplex DNA molecules (Figure 1.2). This requirement of DNA polymerases to use primers to initiate DNA synthesis is critical for the PCR process since it means we can control where the primers bind, and therefore which region of DNA will be replicated and amplified. If the DNA polymerase was like an RNA polymerase that does not require a primer then we would have no way of defining what segment of DNA we wanted to be copied.
At the next heating step the double-stranded molecules, which are heteroduplexes containing an original template DNA strand and a newly synthesized DNA strand produced during the first DNA synthesis reaction, are now denatured. Each DNA single strand can now act as a template for the next round of DNA synthesis. As discussed in detail in Chapter 2, it is during this second cycle of PCR that the first DNA single strand of a length defined by the positions of the primers can be formed. In cycle 3 the first correct length double-stranded PCR products are formed. In subsequent cycles there is then an exponential increase in the number of copies of the ‘target’ DNA sequence; theoretically, the number of copies of the target sequence will be doubled at each PCR cycle. This means that at 100% efficiency, each template present at the start of the reaction would give rise to 106 new strands after only 20 cycles of PCR. Of course the process is not 100% efficient, and it is usually necessary to carry out more reaction cycles, often 25 to 40 depending upon the concentration of the initial template DNA, its purity, the precise conditions and the application for which you require the product. The specificity and efficiency of PCR, however, means that very low numbers of template molecules present at the start of the PCR can be amplified into a large amount of product DNA, often a microgram or more, which is plenty for a range of detailed analyses. Of course, this ability to amplify also means that if you happen to contaminate your reaction with a few molecules of product DNA from a previous reaction, you may get a false result. This is why performing control reactions is so important and we will deal with such contamination problems in Chapter 4.
1.3 PCR is controlled by heating and cooling
PCR relies on the use of different temperatures for the three steps of the reaction, denaturation, annealing and extension. A high temperature, usually 94–95°C, is used to denature (separate) the strands of the DNA template. The temperature is then lowered to allow the primers to anneal by base pairing to their complementary sequences on the template strands; this temperature varies depending on the primers (see details in Chapter 3).
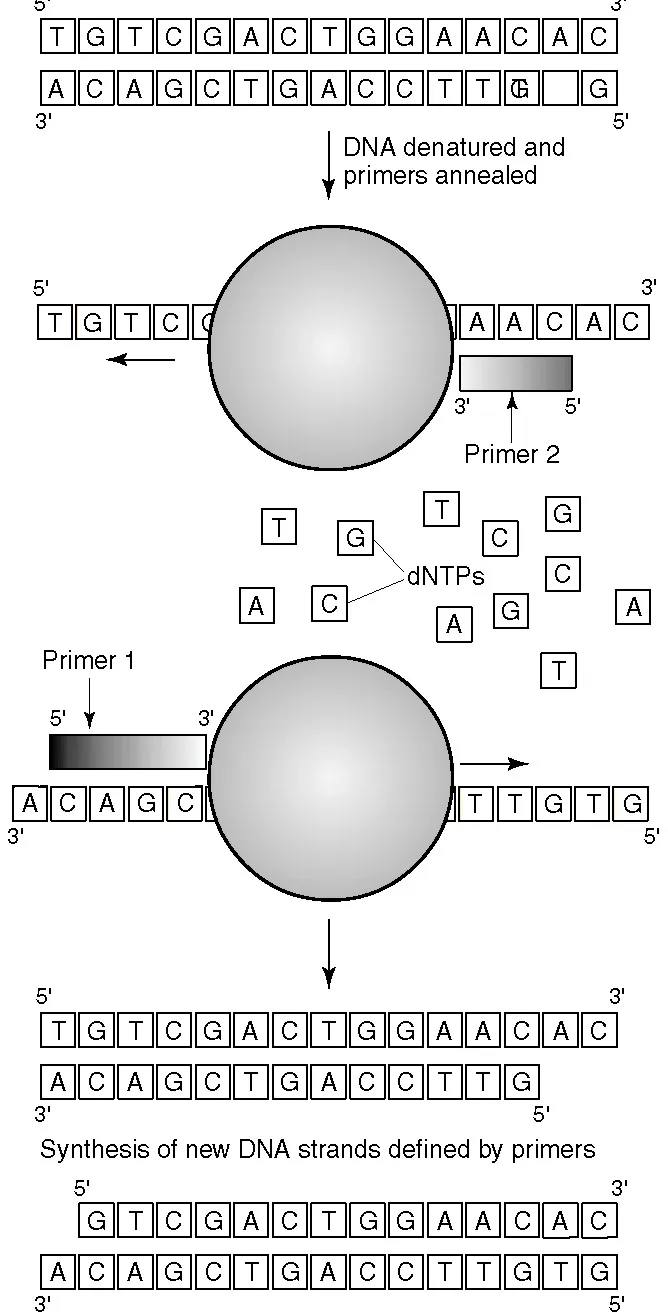
Figure 1.2
The first cycle of a PCR. A double-stranded template molecule is denatured. Primers anneal to their complementary sequences on the single-stranded template. DNA synthesis is catalyzed by a thermostable DNA polymerase. The result of this PCR cycle is that two copies of the target sequence have been generated for each original copy.
The annealing temperature is important to ensure high specificity in the reaction; generally the higher the annealing temperature the more specific will be the reaction. A temperature of 55°C is commonly used, but in many cases a higher temperature is better and this can even be as high as 72°C for some experiments, leading to a two-temperature PCR cycle. Finally, for efficient DNA synthesis, the temperature is adjusted to be optimal for the DNA polymerase activity, normally 72°C (see Chapter 3). To amplify the target DNA it is necessary to cycle through these temperatures several times (25 to 40 depending on the application). Conveniently, this temperature cycling is accomplished by using a thermal cycler, a programmable instrument that can rapidly alter temperature and hold samples at the desired temperature for a set time. This automation is one of the important advances that led to PCR becoming widely accessible to many scientists and is covered in more detail in Chapter 3. Before thermal cyclers became available, PCR was performed by using three water baths set to temperatures of typically 95°C, 55°C and 72°C, and reaction tubes in racks were moved manually between the baths.
The other major technological advance that preceded the development of thermal cyclers was the replacement of DNA polymerase I Klenow fragment with thermostable DNA polymerases, such as Taq DNA poly-merase, which are not inactivated at the high denaturation temperatures used during PCR. The ability to carry out the reaction at high temperatures enhances the specificity of the reaction (Chapter 4). At 37°C, where Klenow works best, primers can bind to nontarget sequences with weak sequence similarity, because mismatches between the two strands can be tolerated. This leads to poor specificity of primer annealing and the amplification of many nontarget products. The introduction of thermostable DNA polymerases also reduced the cost of a reaction by reducing the amount of polymerase required. With Klenow, at each denaturing step the enzyme was also denatured and therefore a fresh aliquot had to be added at each cycle. Thermostable polymerases retain their activity at the denaturation temperatures and therefore only need to be added at the start of the reaction.
1.4 PCR applications and gene cloning
PCR has revolutionized our approach to basic scientific and medical research, to medical, forensic and environmental testing. It provides an extremely flexible tool for the research scientist, and every molecular biology research laboratory now uses PCR routinely; often adapting and tailoring the basic procedures to meet their own special needs. It has become an indispensable tool for routine and repetitive DNA analyses such as diagnosis of certain genetic diseases within clinical screening laboratories where speed and accuracy are important factors, and also for sample identification in forensic and environmental testing. In particular PCR has become a central tool in the analysis and exploitation of genome sequence information, for example in gene knockout through RNA interference where PCR allows the rapid generation of appropriate constructs. It also facilitates measurement of levels of gene expression by ‘real-time’ PCR that monitors the level of product amplification at each cycle of the PCR (Chapter 9), providing information on the relative concentrations of template cDNA.
In some cases PCR provides an alternative to gene cloning, but in other cases it provides a complementary tool. In gene cloning a fragment of DNA is joined by ligation to a cloning vector which is able to replicate within a host cell such as the bacterium Escherichia coli. As the bacterium grows, the new recombinant DNA molecule is copied by DNA replication, and as the cell divides the number of cells carrying the recombinant molecule increases. Finally, when there are enough cells you can isolate the recombinant DNA molecules to provide sufficient DNA for analysis or further manipulation of the cloned DNA fragment. This type of cloning experiment takes about 2–3 days. PCR also amplifies your target DNA fragment so that you have enough to analyze or manipulate, but in this case the DNA replication occurs in a test-tube and usually takes no more than 1–3 hours. In many cases, for example in diagnostic tests for cancers or genetic diseases, including antenatal screening, or in forensic testing, PCR provides the most sensitive and appropriate approach to analyze DNA within a day.
For studying new genes and genetic diseases it is often necessary to create gene libraries and this may involve PCR followed by cloning into a suitable vector. Also many experiments to produce proteins to study their structure and function require expression in host cells and this requires the cloning of the gene perhaps as a PCR product, into a suitable expression vector. So in many cases PCR and gene cloning represent complementary techniques.
It is important to consider carefully the most appropriate strategy for the...
It is important to consider carefully the most appropriate strategy for the...
Table of contents
- Cover Page
- Title Page
- Copyright Page
- Abbreviations
- Preface
- 1. An Introduction to PCR
- 2. Understanding PCR
- 3. Reagents andInstrumentation
- 4. Optimization of PCR
- 5. Analysis, Sequencing andIn Vitro Expression of PCRProducts
- 6. Purification and Cloningof PCR Products
- 7. PCR Mutagenesis
- 8. Analysis of GeneExpression
- 9. Real-Time RT-PCR
- 10. Cloning Genes By PCR
- 11. Genome Analysis
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.4M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access PCR by Mike McPherson,Simon Møller in PDF and/or ePUB format, as well as other popular books in Biological Sciences & Biology. We have over one million books available in our catalogue for you to explore.