This handbook provides clinical guidance to the practicing physician on the diagnosis and treatment of Interstitial Lung Diseases (ILD). A contributed work with invited chapters which draw on the knowledge and experience of recognised global leaders in respiratory medicine, it is authoritative, concise and portable and is intended for use in a fast-paced clinical setting. The book:
- offers practical tips and clear guidance for clinicians
- provides detailed explanations of the main therapeutic options for each individual ILD
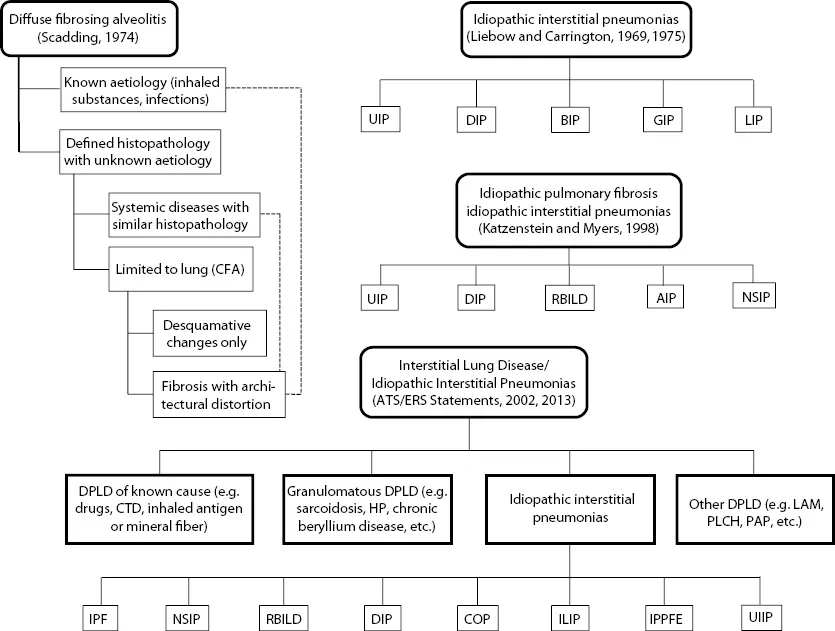
- contains high-quality visuals, including radiology and histopathology of the most common as well as some of the rarer ILDs
- discusses individual ILDs and has topics common to all including critical care, lung transplantation and palliative care
- navigates clinicians through cases with decision making guidelines and algorithms
- includes appendices with international practice guidelines, sample patient information sheets and other helpful resources.
Emphasizing how to perform a thorough assessment of an ILD patient for accurate diagnosis and their subsequent effective management, this is both a gold standard text as well as a daily companion for physicians caring for ILD patients. A first-of-its-kind, it will become the go-to guide for all clinicians who manage patients with ILD.