![]()
PART I
Inherited renal disorders
41 Tubulopathies
Detlef Bockenhauer
42 Renal tubular acidosis
John W. Foreman
43 Cystic kidney disease
Lisa M. Guay-Woodford
44 Ciliopathies and nephronophthisis
John Sayer, Shreya Raman, and Shalabh Srivastava
45 Alport syndrome and thin basement membrane nephropathy
Michelle N. Rheault and Clifford E. Kashtan
46 Renal disease in syndromic disorders
Patrick Niaudet
![]()
41
Tubulopathies
DETLEF BOCKENHAUER
Organization of tubules
Disorders of the proximal tubule
Disorders of thick ascending limb of loop of henle
Disorders of distal convoluted tubule
Disorders of collecting duct
References
Review questions
Answer key
Assuming a glomerular filtration rate of 100 mL/min, the kidneys of an average adult each day filter approximately 150 L of plasma each day, containing approximately 20 mol of sodium (Na+), the equivalent of more than a kilogram of table salt. Thus, without tubules, we all would have to drink 150 L a day and eat a kilogram of salt to keep up with urinary losses. The enormity of these numbers demonstrates the importance of proper tubular function. Typically, more than 99% of filtered salt and water are reabsorbed. Yet, the tubules not only need to accomplish the massive bulk transport of water and solutes, they also need to finely regulate the precise amount of reabsorption to balance variable intake and extrarenal losses, to maintain volume and electrolyte homoeostasis.
ORGANIZATION OF TUBULES
The enormous feat of complex tubular functions is accomplished by a whole orchestra of tubular transporters arranged along the entire nephron. Each transporter plays a specific role in coordinating a portion of tubular function. Much has been learned about the individual transport processes through observations of patients in whom one or more of these transport processes is defective, or patients with tubulopathies.
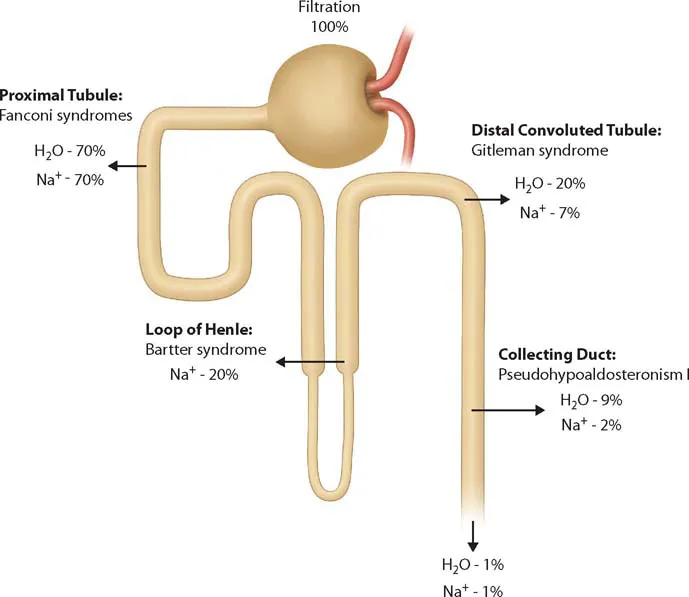
Both from a functional and a histoanatomic perspective, the nephron can be divided into four different segments (Figure 41.1):
Proximal tubule (PT)
Loop of Henle
Distal convoluted and connecting tubule (DCT)
Collecting duct (CD)
Disorders of the loop of Henle are mainly restricted to the thick ascending limb (TAL). In general, high-capacity bulk transport occurs in the PT, whereas the fine tuning of reabsorption occurs mostly in the distal end, the CD. Disorders of each segment tend to have a distinctive “fingerprint” of clinical and biochemical abnormalities that informs the diagnosis (Table 41.1).
KEY POINT
The renal tubule can be separated morphologically and functionally into four main segments: PT, loop of Henle, DCT, and CD.
DISORDERS OF THE PROXIMAL TUBULE
PHYSIOLOGY
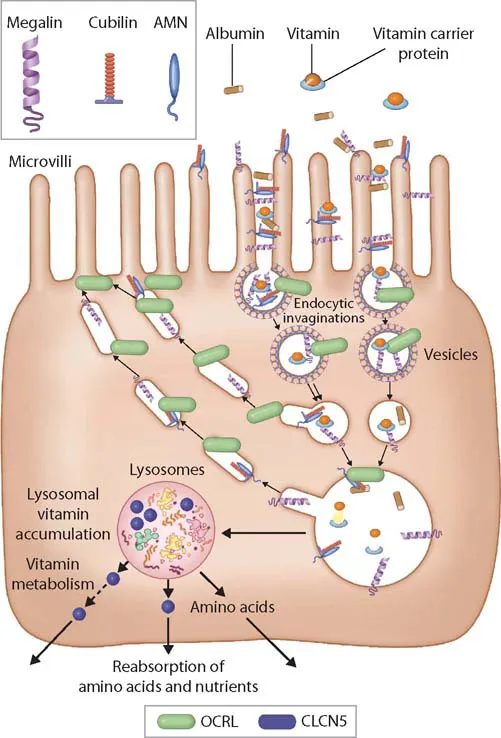
The PT is not only the site of high-capacity transport, but it is also the exclusive site of reabsorption for several solutes, such as low-molecular-weight (LMW) proteins, amino and organic acids, glucose, and phosphate.1 Because a sizeable proportion of albumin is also filtered, PT dysfunction is associated with moderately elevated urinary albumin levels, which can lead to confusion with glomerular disorders.2 Albumin, like LMW proteins and a host of other solutes, is taken up by the proximal tubular epithelial cells through the promiscuous receptors megalin and cubilin, which are then internalized with the cargo into endosomes (Figure 41.2).3
Defects in PT can range from very specific dysfunction, such as isolated renal glucosuria, caused by impaired function of the Na+-glucose cotransporter SGLT2 (SLC5A2),4 to a generalized dysfunction of the PT. Clinically, cases of generalized PT dysfunction with glucosuria, acidosis, and hypophosphatemic rickets were first described in the 1930s by De Toni,5 Debre et al.,6 and Fanconi,7 and consequently this constellation of symptoms is referred to as De Toni– Debre-Fanconi syndrome, or just renal Fanconi syndrome. It can be acquired or inherited, either isolated or in the context of systemic disorders, such as mitochondrial cytopathies or nephropathic cystinosis (Table 41.2).8
CYSTINOSIS
Cystinosis, albeit a rare disorder with an estimated incidence of approximately less than 1 in 100,000, is the most common inherited cause of renal Fanconi syndrome in children.9 The underlying gene, CTNS, encodes a transporter involved in exporting cysteine from lysosomes.10 Consequently, impaired CTNS function leads to intracellular cystine accumulation.11 However, how exactly this accumulation leads to cellular damage has yet to be clarified, although several hypothesis have been proposed, such as increased apoptosis, impaired energy production, and altered glutathione metabolism.12
Clinical features and diagnosis
Although cystinosis is a multiorgan disorder, the renal manifestations initially dominate, potentially because of the high cystine turnover in the PT from amino acid and LMW protein reabsorption. Measurement of urinary excretion of several LMW proteins, such as α1- and β2-microglobulins, can be used as markers for LMW proteinuria, but retinol-binding protein has been shown to have the highest sensitivity and specificity13 for proximal tubular proteinuria.
Presentation is usually in the first 2 years of life with poly-uria or polydipsia, failure to thrive, and rickets. Biochemical testing reveals the typical features of renal Fanconi syndrome (Table 41.3). However, because cystinosis affects most cells, other renal segments can be affected: glomerular function deteriorates with time, and other tubular features, such as secondary nephrogenic diabetes insipidus, have also been reported.14
Table 41.1 Selected tubulopath¡es, associated genes, and pertinent clinical and biochemical characteristics*
Once suspected, the diagnosis is easily established by slit-lamp examination of the eyes, which demonstrates characteristic cystine crystals in virtually all children older than 16 months of age.15 The absence of crystals before this age does not exclude the diagnosis. The diagnosis can be confirmed by measurement of an increased leukocyte cystine content or genetic testing, or both. Most children with typical clinical features of cystinosis have identifiable biallelic mutations in CTNS.16
Later in life, other types of organ dysfunction can become apparent, such as hypothyroidism, myopathy with swallowing dysfunction, retinal blindness, diabetes mellitus, male hypogonadism, and pulmonary and central nervous system dysfunction.9
Table 41.2 Causes of renal Fanconi syndrome
Treatment and prognosis
Treatment used to be just supportive, by replacing renal fluid and solute losses, enhancing nutrition, and treating the rickets. Patients developed end-stage renal disease (ESRD) i...