![]()
1 Introduction to Drug Design and Discovery
Ulf Madsen and Povl Krogsgaard-Larsen
CONTENTS
1.1 Medicinal Chemistry: An Interdisciplinary Science
1.2 Drug Discovery: A Historical Perspective
1.3 Drug Development Process: An Outline
1.4 Discovery of Drug Candidates
1.4.1 Natural Products: Role in Target Identification
1.4.2 Natural Products as Lead Structures
1.4.3 Basic Principles in Lead Development and Optimization
1.4.3.1 Bioisosteres
1.4.3.2 Stereochemistry
1.4.3.3 Membrane Penetration: Including the Lipinski Rule of Five
1.4.3.4 Structure-Based Drug Design
1.5 Individualized Medicine and Concluding Remarks
Further Reading
1.1 MEDICINAL CHEMISTRY: AN INTERDISCIPLINARY SCIENCE
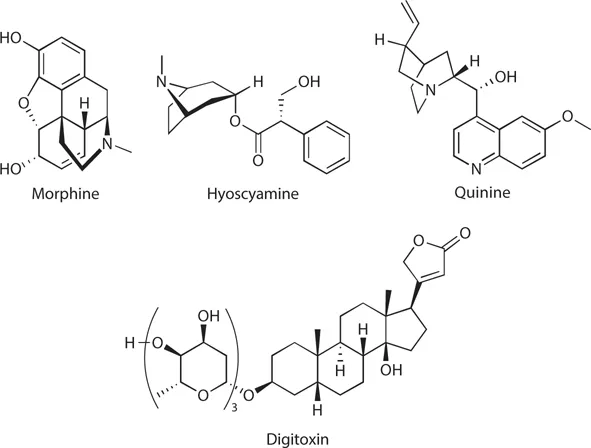
Therapeutic agents are chemical entities that prevent disease, assist in restoring health to the diseased, or alleviate symptoms associated with disease conditions. Medicinal chemistry is the scientific discipline that makes such drugs available either through discovery or design processes. Throughout history, drugs were primarily discovered by empirical methods, investigating substances or preparations of materials, such as plant parts or plant extracts, found in the local environment. Over the previous centuries, chemists developed methods for the isolation and purification of the active principles in medicinal plants. The purification and structure determination of natural products like morphine, hyoscyamine, quinine, and digitalis glycosides represent milestones in the field of drug discovery and the beginning of medicinal chemistry as a fascinating independent field of research (Figure 1.1).
In the twentieth century, a very large number of biologically active natural products were structurally modified in order to optimize their pharmacology and drug properties in general, and novel drugs were prepared by an increasing use of advanced synthetic methods. Moreover, the rapidly growing understanding of the nature of disease mechanisms, how cells function, and how drugs interact with cellular processes has led to the rational design, synthesis, and pharmacological evaluation of new drug candidates. Most recently, new dimensions and opportunities have emerged from a deeper understanding of cell biology, genetics, and biostructures.
Modern medicinal chemistry draws upon many scientific disciplines, with organic chemistry, physical chemistry, and pharmacology being of fundamental importance. But other disciplines such as biochemistry, molecular biology, toxicology, genetics, cell biology, biophysics, physiology, pathology, and computer modeling approaches play important roles. The key research objective of medicinal chemistry is to investigate relationships between chemical structure and biological effects. When the chemical structure of a particular drug candidate has been optimized to interact with the biological target, the compound further has to fulfill a multifaceted set of criteria before it can be safely administered to patients. Absorption, distribution, metabolism, excretion (ADME), and toxicology studies in animals and humans are time-consuming research tasks which often call for redesign of the chemical structure of the potential therapeutic agent investigated. It is an iterative process which in reality ends up in an overall compromise with respect to multiple desired properties.
1.2 DRUG DISCOVERY: A HISTORICAL PERSPECTIVE
In early times, there was no possibility of understanding the biological origin of a disease. Of necessity, progress in combating disease was disjointed and empirical. The use of opium, ephedra, marijuana, alcohol, salicylic acid, digitalis, coca, quinine, and a host of other drugs still in use long predate the rise of modern medicine. These natural products are surely not biosynthesized by plants for our therapeutic convenience, but they normally have survival value to the plants in dealing with their own ecological challenges.
The presence of biologically active substances in nature, notably in certain plants, was in medieval times interpreted more teleologically. In the early sixteenth century, the Swiss-Austrian medical doctor and natural scientist Paracelsus formulated the “Doctrine of Signatures”:
Just as women can be recognized and appraised on the basis of their shape; drugs can easily be identified by appearance. God has created all diseases, and he also has created an agent or a drug for every disease. They can be found everywhere in nature, because nature is the universal pharmacy. God is the highest ranking pharmacist.
The formulation of this doctrine was in perfect agreement with the dominating philosophies at that time, and it had a major impact on the use of natural medicines. Even today, remanences of this doctrine can be observed in countries where herbal medical preparations are still widely used. Although the “Doctrine of Signatures” evidently is out of the conception of modern medicinal natural product research, the ideas of Paracelsus were the first approach to rational drug discovery.
More than 100 years ago, the mystery of why only certain molecules produced a specific therapeutic response was rationalized by the ideas of Emil Fischer and further elaborated by John Langley and Paul Ehrlich that only certain cells contained receptor molecules that served as hosts for the drugs. The resulting combination of drug and receptor created a new super molecule that had properties producing a response of therapeutic value. One extension of this conception was that the drug fits the target specifically and productively like “a key into its corresponding lock.” When the fit was successful, a positive pharmacological action (agonistic) followed, analogous to opening the door. On the contrary, when the fit prevented the intrinsic key to be inserted an antagonist action resulted—i.e., the imaginative door could not be opened. Thus, if one had found adventitiously a ligand for a receptor, one could refine its fit by opportunistic or systematic modifications of the ligand’s chemical structure until the desired function was obtained.
This productive idea hardly changed for the next half century and assisted in the development of many useful drugs. However, a less fortunate corollary was that it led to some limitations of creativity in drug design. The drug and its receptor (whose molecular nature was unknown when the theory was formulated) were each believed to be rigid molecules precrafted to fit one another precisely. Today, we know that receptors are highly flexible transmembrane glycoproteins accessible from the cell surface that often comprise more than one drug compatible region. Further complexities have been uncovered continually. For example, a number of receptors have been shown to consist of clusters of proteins either preassembled or assembled as a consequence of ligand binding. The component macromolecules may be either homo- or heterocomplexes. The challenge of developing specific ligands for systems of this complexity may readily be imagined (Chapters 4 and 12).
The opposite extreme to “the lock and key model” is “the zipper model.” In this view, a docking interaction takes place (much as the end of a zipper joins the talon piece) and, if satisfactory complementarity is present, the two molecules progressively wrap around each other and adapt to the steric and electrostatic needs of each other. A consequence of accepting this mutual adaptation is that knowledge of the receptor ground state may not be particularly helpful as it adjusts its conformation to ligand binding. Thus, in many cases, one now tries to determine the 3D structure of the receptor–ligand complex. In those cases where X-ray analysis remains elusive, modeling of the interactions involved is appropriate. This is the subject of Chapters 2, 3 and 4.
Earlier, it was also noted that enzymes could be modulated for therapeutic benefit. Enzymatic proteins share many characteristics with receptors, although enzymes catalyze biochemical reactions. Receptor ligands interact with the receptor glycoproteins or with the interfaces between the macromolecular subunits of di- or polycomponent receptor complexes and modify the conformation and dynamics of these complexes. Thus, neither receptor agonists nor antagonists directly interfere with chemical reactions and generally are dissociated from the receptor recognition sites structurally unchanged.
The reaction mechanisms underlying the function of the vast majority of enzymes have been elucidated in detail, and based on such mechanistic information, it has been possible to design a variety of mechanism-based enzyme inhibitors, notably kcat inactivators and transition-state analogs, many of which are in therapeutic use (Chapter 11). Until very recently, it was usually only possible to inhibit enzyme action rather than facilitate it. Actually, diseases frequently result from excessive enzymatic action, making selective inhibition of these enzymes therapeutically useful.
Much later, further classes of receptors were disclosed, explored, and exploited as therapeutically relevant pharmacological targets. This heterogeneous group of receptors comprises nuclear receptors operated by steroid hormones and other lipophilic biochemical mediators, a broad range of membrane-ion channels (Chapter 13), DNA or RNA (Chapter 22), and a number of other biostructures of known or unknown functions. These aspects will be discussed in different chapters of this book.
1.3 DRUG DEVELOPMENT PROCESS: AN OUTLINE
The stages through which a drug discovery/development project proceeds from inception to marketing and beyond are illustrated in Figure 1.2 and described briefly in the following text. The discovery and development process can be described by a number of individual steps, but is also a continuous and iterative process not necessarily performed in a strict stepwise process. From this outline, the complexity of the task of finding new therapeutic agents is evident:
Target discovery comprises identification and validation of disease-modifying targets. Two major strategies are used for target identification and validation: (1) the molecular approach, with focus on the cells or cell components implicated in the disease and the use of clinical samples and cell models, and (2) the systems approach based on target discovery through the study of diseases in whole organisms.
Before or after identification of target disease, establishment of a multidisciplinary research team, selection of a promising approach, and decision on a sufficient budget. Initiation of chemistry normally involves synthesis based on available chemicals, in-house chemical libraries, or collection of natural product sources. Start of pharmacology includes suitable screening methods and choice of receptor or enzymatic assays.
Confirmation of potential utility of initial class(es) of compounds in animals, focusing on potency, selectivity, and apparent toxicity.
Analog syntheses of the most active compounds, planned after careful examination of literature and patents. More elaborated pharmacology in order to elucidate the mode of action, efficacy, acute and chronic toxicity, and genotoxicity. Studies of ADME characteristics. Planning of large-scale synthesis and initiation of formulation studies. Application for patent protection.
These first project phases which typically last 4–5 years, are followed by highly time- and resource-demanding clinical, regulatory, and marketing phases which normally last 6–10 years: