An authoritative introduction to Parkinson's disease and its related disorders and syndromes, this book provides a concise overview of the disease and its diagnosis and management. The author presents samples of clinical, investigative (CT, MRI, and PET), and pathological images with succinct descriptive text of the disorders featured. He describes

- 80 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
An Illustrated Pocketbook of Parkinson's Disease and Related Disorders
About this book
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Related disorders
Progressive supranuclear palsy (Steele-RichardsonOlszewski syndrome)
For many, or perhaps even all, patients with extrapyramidal syndromes, a classical picture has been described which is anticipated to predict a particular pathological entity at postmortem examination. As knowledge of the disease grows, however, it soon becomes apparent that the same disease process—as defined pathologically—has a much broader clinical spectrum than was appreciated in the original description. The converse also applies: patients with a classical clinical syndrome may prove to have other pathological entities.
Nowhere are these discrepancies more evident than in cases of progressive supranuclear palsy (PSP). One of the problems in establishing clinicopathological correlations in PSP is the lack of consensus as to the pathological criteria for the diagnosis. Certain features, however, are predictable. The substantia nigra shows severe pigment depletion, as does the locus ceruleus. Neuronal loss is found in the substantia nigra, subthalamus, and globus pallidus. Neurofibrillary tangles can be identified in the cerebral cortex, caudate, putamen, globus pallidus, subthalamus, and brain stem (Figure 26). Accompanying the neurofibrillary tangles are neuropil threads (silverand tau-positive). Typically, changes are found in the regions associated with vertical gaze, including the rostral interstitial nucleus of the medial longitudinal fasciculus and the interstitial nucleus of Cajal Tau is a microtubule-associated protein which is closely involved in neuronal axonal transport. Frequent tau-positive inclusions are found in PSP and corticobasal degeneration
A disturbance of gait is common in PSP and many patients are liable to falls. The body tends to remain extended rather than taking on the stooped posture of Parkinson's disease. Pseudobulbar features are prominent, with dysphagia, dysarthria, and emotional incontinence. The supranuclear palsy first affects down gaze, and particularly downward saccades (Figure 27). Some patients complain of blurred vision or frank diplopia. Later, vertical, then horizontal, saccades become compromised, followed by impairment of pursuit movement. Reflex eye movements, elicited by the doll's head maneuver, are spared initially (Figure 28), but are later lost so that a total ophthalmoplegia becomes evident. In well-documented cases, despite the appropriate pathological changes found at postmortem, the patient may have had no disturbances of eye movements in life. Limb rigidity is less prominent than axial rigidity. Bradykinesia is present to a varying degree, with some patients presenting as a pure akinetic syndrome. Tremor occurs in around 12–16% of cases. A subcortical, rather than cortical, dementia is characteristic, but, in the later stages, a dementia of frontal type with memory impairment, dysphasia and apraxia is common,
In most cases, dopa therapy is ineffective in PSP and almost never influences the ophthalmoplegia.
Imaging changes in PSP include both generalized and selective brain stem atrophy (Figure 29). Single photon emission computed tomography (SPECT) can demonstrate impairment of frontal perfusion with an intact cortical rim. PET scanning shows decreased metabolic activity in the frontal cortex, caudate and putamen, together with evidence of abnormal D2 receptor function (Figure 30).
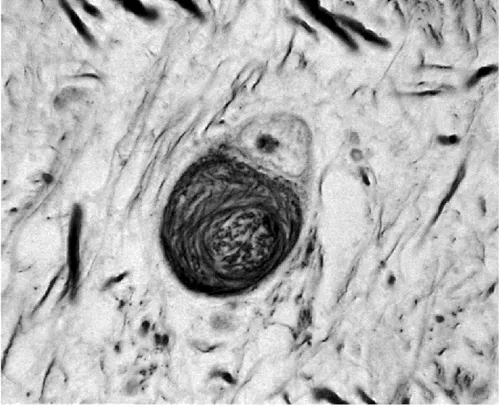
Figure 26 Progressive supranuclear palsy: subthalamic neurons showing neurofibrillary tangle (Bielschowsky silver impregnation)
Striatonigral degeneration
This condition is frequently confused with Parkinson's disease in life. At postmortem, there are atrophy and discoloration of the putamina (Figure 31) accompanied, in almost half of the cases, by atrophy of the caudate nuclei. The changes in the putamen begin dorsally in the posterior two-thirds, then spread ventrally and anteriorly. On microscopy, the putamen shows intracellular pigmentation, gliosis and loss of myelinated fibers (Figure 32). Neuronal depletion, gliosis and loss of myelinated fibers are seen in the globus pallidus, whereas both the substantia nigra and locus ceruleus show pallor with microscopic evidence of neuronal loss and gliosis (Figures 33 and 34). Lewy bodies are seldom found. In some cases, even without clinical features in life, there is involvement of the olivopontocerebellar system. Striatonigral degeneration has considerable clinical overlap with Parkinson's disease, but sufficient differences to suggest the diagnosis in life. Rest tremor in the early stages of the disease is distinctly uncommon, although it appears in half of the cases during the later stages of the disease. The condition is equally likely as Parkinson's disease to be asymmetrical at onset. Falls early in the course of the disease are a recognized feature. Other features which should suggest the diagnosis include severe dysphonia and dysphagia, and the development of autonomic symptoms or cerebellar signs, indicating the development of multiple system atrophy (vide infra).
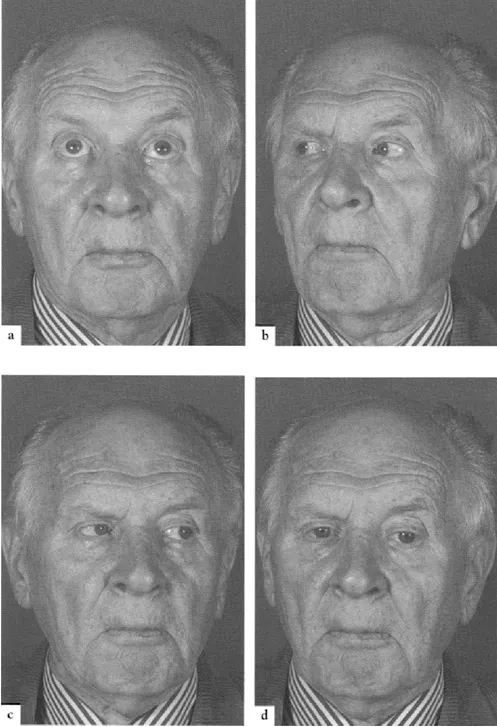
Figure 27 Progressive supranuclear palsy: upward (a), lateral (b and c) and down (d) gaze
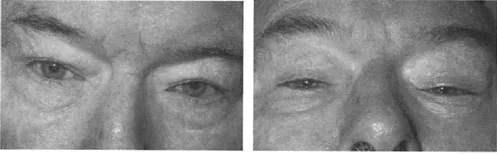
Figure 28 Progressive supranuclear palsy: defective doll's head maneuver on down gaze
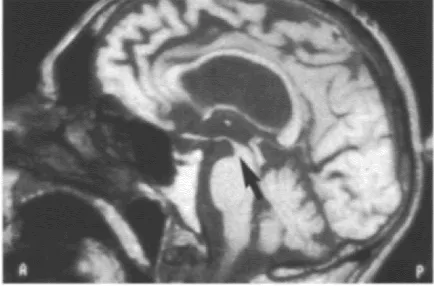
Figure 29 Progressive supranuclear palsy: sagittal T1-weighted MRI showing midbrain atrophy. Courtesy of M.Savoiardo, Department of Neuroradiology, Istituto Nazionale Neurologico ‘C.Besta’, Milan, Italy
Multiple system atrophy
Autonomic features may accompany a parkinsonian syndrome without evidence of other system involvement. In such patients, the autonomic failure is due to intermediolateral column degeneration in the spinal cord, whereas the parkinsonian syndrome reflects the classical features of idiopathic Parkinson's disease, including typical changes in the substantia nigra and locus ceruleus, with Lewy body formation. In other patients described as having multiple system atrophy, the autonomic failure is due to the same pathological process in the spinal cord, but the other clinical features represent a combination, in varying degrees, of striatonigral degeneration and olivopontocerebellar atrophy (OPCA).
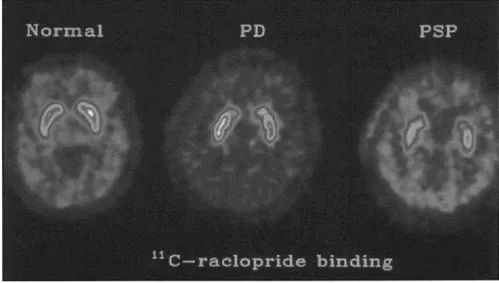
Figure 30 [11C] Raclopride binding in normal subject (left) compared with a parkinsonian patient (middle) and a patient with PSP (right)
In OPCA, there is macroscopic evidence of atrophy of the pons, middle cerebellar peduncle, parts of the cerebellum and the olives (Figure 35). Microscopically, the pontine tegmentum is virtually spared, but there is pallor of the transverse fibers in the basis pontis, together with neuronal loss (Figure 36). Depletion of both granules and Purkinje cells is seen in the cerebellum. Where the latter has occurred, empty ‘baskets’ with hypertrophied fibers are seen associated with the formation of axon ‘torpedoes’ in the molecular layer (Figure 37). Oligodendroglial cytoplasmic inclusions are probably seen in all cases of multiple system atrophy and in all sporadic cases of OPCA, but only rarely in familial cases of OPCA (Figure 38). The inclusions stain positive for synuclein.
Clinical criteria have been suggested for the diagnosis of multiple system atrophy (Table 3). Diagnostic problems arise as the result of some patients presenting with parkinsonism, others with a cerebellar syndrome, and a third group with autonomic failure, without clear evidence in all three instances of other system involvement. Sporadic cases are not seen in those under 30 years of age. Dementia is not a feature of multiple system atrophy, nor is there an ophthalmoplegia (although this is recorded in both sporadic and familial forms of OPCA). Although poor or absent dopa responsiveness is the norm, some cases— confirmed at postmortem examination—may show a response comparable to that seen in idiopathic Parkinson's disease.
Multiple system atrophy usually presents in the sixth decade of life. The median survival is of the order of 7–8 years. Men are slightly more often affected than women. The most common combination of clinical features is autono...
Table of contents
- Cover Page
- Title Page
- Copyright Page
- Anatomy
- Parkinson's disease
- Parkinsonian syndromes
- Related disorders
- Selected bibliography
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access An Illustrated Pocketbook of Parkinson's Disease and Related Disorders by G. David Perkin in PDF and/or ePUB format, as well as other popular books in Medicine & Medical Theory, Practice & Reference. We have over 1.5 million books available in our catalogue for you to explore.