
eBook - ePub
Preparing for FDA Pre-Approval Inspections
A Guide to Regulatory Success, Second Edition
- 304 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Preparing for FDA Pre-Approval Inspections
A Guide to Regulatory Success, Second Edition
About this book
This Second Edition is an essential guide to preparing for FDA pre-approval inspections-taking into account current trends in FDA expectations and inspection activities, such as the GMPs of the 21st Century, quality systems-based approach to inspections, risk-based inspections, quality by design, process analytical technology, design space, etc. Th
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Subtopic
Administrative LawIndex
Business1
The Evolution of the Food and Drug Administration: Pre–New Drug Application Approval Inspection
Martin D. Hynes and Jeanette M. Buckwalter
Eli Lilly and Company, Indianapolis, Indiana, U.S.A.
A HISTORICAL OVERVIEW: PAST, PRESENT, AND FUTURE
The Food and Drug Administration’s (FDA) pre-approval inspections (PAIs) program is an investigation by the agency to review the adequacy and accuracy of the information provided in a regulatory submission, most frequently the New Drug Application (NDA). This program was first implemented in the FDA’s mid-Atlantic region in the late 1980s. Formal communication of the program came in 1990 with a publication entitled “Mid-Atlantic Region Pharmaceutical Inspection Program” authored by Henry Avallone (1). Shortly thereafter, the FDA issued a formal compliance manual entitled “The FDA Compliance Program Guidance Manual (CPGM) on Pre-Approval Inspections/Investigations (Program 7346.832)” (2). The manual, issued in October 1990, outlined a role for both the Center for Drug Evaluation and Research (CDER) and the district offices in the drug approval process, thus adding the new step of a compliance review to the approval process. The application for marketing approval was now to be reviewed for good manufacturing practices (GMPs) compliance as well as the adequacy and accuracy of the data it included. Prior to this time, companies were on the honor system, with the FDA trusting sponsoring companies to submit accurate data in their marketing application. The advent of the generic drug scandal in the late 1980s brought an end to the honor system, which was replaced by the need to audit all of the data submitted to the FDA for adequacy and accuracy prior to approval of the application (3).
In addition to this historical perspective, these inspections can also be understood within the context of the entire product development process and the myriad of compliance regulations that govern it.
The new product development process for a pharmaceutical begins with discovery and ends when, for all intents and purposes, the drug product is no longer made available to patients in the global marketplace. Numerous scientists and technical experts carry out the product development work, which begins with discovery activities. It is a lengthy and expensive process. This development work results in the delivery of two major classes of “products.” The first of these products is the drug product or the material itself, while the second product is information about the new drug product that has been generated during the entire drug development process.
The material that is generated during the drug development process is utilized for a variety of purposes, including preclinical studies, within product development, discovery, as well as in the toxicology functions, and for clinical trials that range from phase I safety studies through phase IV post-marketing surveillance. Given the intended uses of this material in preclinical safety studies, the development process must meet the regulatory requirements that are outlined in the Good Laboratory Practices (GLPs) (4,5), which include documenting synthesis methods and maintaining adequate batch and stability records. The material that is produced for use in clinical trials must meet the regulatory requirements that are prescribed in the Good Clinical Practices (GCPs) (6,7) as well as be manufactured in accordance with the GMPs (8). The GCPs require drug accountability and record keeping, and that material used in clinical trials is manufactured under the GMPs. The 1991 Investigational New Drug (IND) guidelines state that “when drug development reaches the stage when the drug products are produced for clinical trials in humans and animals, then compliance with the GMPs is required” (9). More recent guidance will be reviewed in the chapter.
The information product or deliverable of the new product development process includes all of the data and information that is generated. Data and information that is generated during the development process is generally captured in lab notebooks as well as in a variety of electronic systems, and then summarized in a series of technical reports. These technical reports are written throughout the course of the development process beginning at the initial stages of product development. Toward the end of the product development process, these detailed technical documents and study reports are utilized to generate at least three major types of summary technical documents: the Development History Report, the Common Technical Document (CTD)/NDA, and the Product Development to Manufacturing Technical Transfer document(s). Only a subset of the data generated during the entire development process is extracted and utilized in one of these three types of technical reports. A schematic representation of this distillation process from technical report to summary report is depicted in Figure 1. As the information depicted in this figure is generated and summarized during the product development process in technical reports, it must be scientifically sound, well documented, accurate, and compliant with all applicable regulatory requirements which would include the GMPs, GCPs, and GLPs. The quality of this information product is assessed at the end of the development process in at least two important ways, first at the time of technology transfer from the product development organization to the manufacturing component and the other at the time of the regulatory review. This transfer can be judged on the basis of a successful transfer of the product development process and analytical control methods from the product development organization to the manufacturing component. A variety of performance indicators can be used to judge the success of this important transfer; they include ease of product and analytical methods validation, number of failed lots, number of issues or problems encountered in the scale up, the time it takes for the transfer to be successful, as well as the number of issues or complaints that are directed to the product development function.
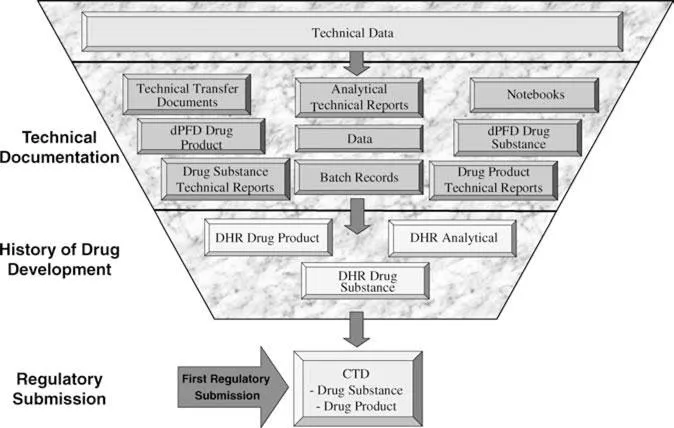
The quality of information produced or generated by the product development organization comes at the time of the regulatory review prior to marketing authorization. This regulatory review includes both a scientific assessment of the information submitted in support of the application as well as a compliance review that occurs during a regulatory inspection like an FDA PAI for marketing authorization. In the United States, the FDA conducts both of these reviews. The scientific review is conducted by CDER, while the compliance review is performed, in large measure by the FDA field compliance groups in concert with other agency groups or functions. (This will be described in greater detail later in the chapter.) These compliance reviews are performed in a general GMP or quality system inspection or through a NDA PAI.
The interrelationship between the drug development process, compliance regulations, and potential regulatory inspections is depicted in Figure 2. As indicated in this figure, it should be noted that in addition to assessments at the time of submission, a compliance assessment of the information or material product from the product development process could literally occur at any time during the entire development process. However for the purpose of this work, we are most concerned with the FDA inspection that occurs at the time of NDA in the United States—the PAI— and hence that will be the focus for the remainder of the chapter. Given this focus, it is important to begin with a definition of what a PAI is.
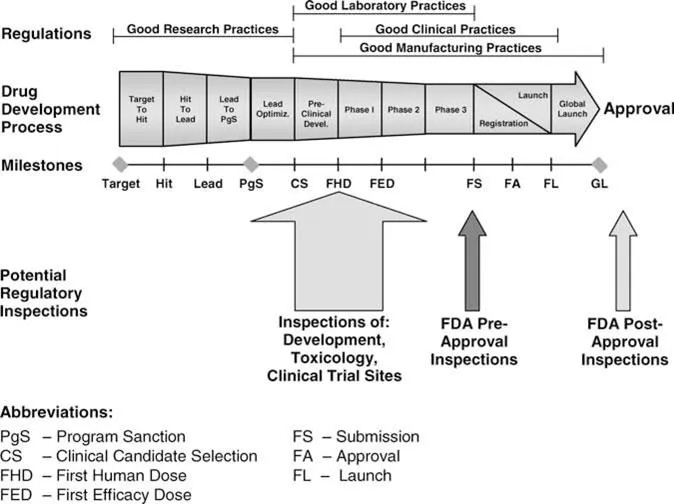
The PAI is an investigation by one or more FDA investigators to review the adequacy and accuracy of the information provided in the regulatory submission. The program is detailed in the FDA CPGM 7346.832 (10). This program was designed by CDER to cover the following types of drugs and companies:
• New chemical entities
• Drugs within a narrow therapeutic range
• Generic versions of the 200 most prescribed drugs
• Drugs that are difficult to manufacture and replicate
• New dosage form for the application
• First approval for the company
• Poor GMP track record by the company
The object of the program is to:
• Ensure that the facilities listed in the application have the capabilities to fulfill the commitments that have been made to the infrastructure and to process, control, package, label, and test the product.
• Ensure that the manufacturing process is validated.
• Ensure that there is a correlation between the manufacturing process for the material utilized in clinical trials, bioavailability studies, and safety studies with the process that was filed with the submission.
• Ensure that the scientific evidence supports full-scale production procedures and controls.
The FDA’s district offices conduct these PAIs. The field investigators from those district offices will audit the data in the submission for both authenticity as well as accuracy. In addition, they will determine the adequacy of the facility, personnel, equipment, and laboratory methods.
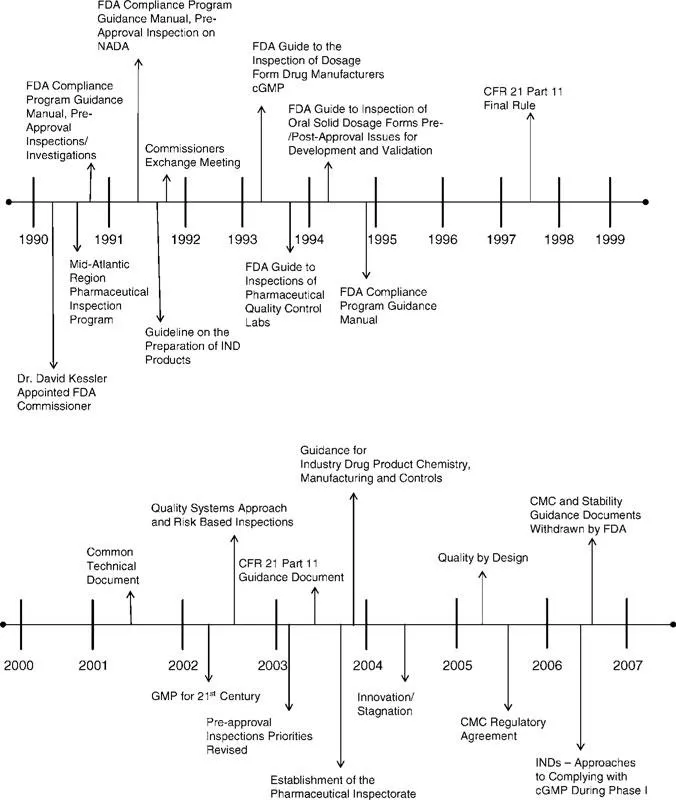
Since the program’s inception in the late 1980s and early 1990s, there has been an ongoing evolution of the FDA PAI program primarily driven by the issuance of new guidance by the FDA. Some of the major changes that have impacted the PAI program, as well as a number of the changes that may alter the program in future, are depicted on the timeline in Figure 3. The remainder of this chapter will provide an overview of this evolution with the intent of providing a snapshot of the program at this point in time and to discuss the potential for future change.
INTRODUCTION TO THE EVOLUTION OF FDA EXPECTATIONS
There are a number of FDA initiatives currently underway that will significantly alter the development, approval, and manufacturing of new pharmaceutical products. The interconnectivity of all of these changes is depicted in Figure 4. It is not possible within the context of this chapter to provide a detailed review of each of these major initiatives. In fact, there are numerous FDA publications, journal articles, and books that cover each of these topics singly in minute detail. However, we will provide a brief overview of some of the most important of these initiatives to give the reader of this work a high level of understanding of these initiatives and their potential impact to pre-NDA approval inspections in the future. The initiatives highlighted here are those that will potentially alter product development and the regulatory review process if implemented, and, therefore, are those that are most likely to impact future PAIs to the greatest extent; hence, this is not meant to be an extensive or exhaustive review of all current regulatory change.
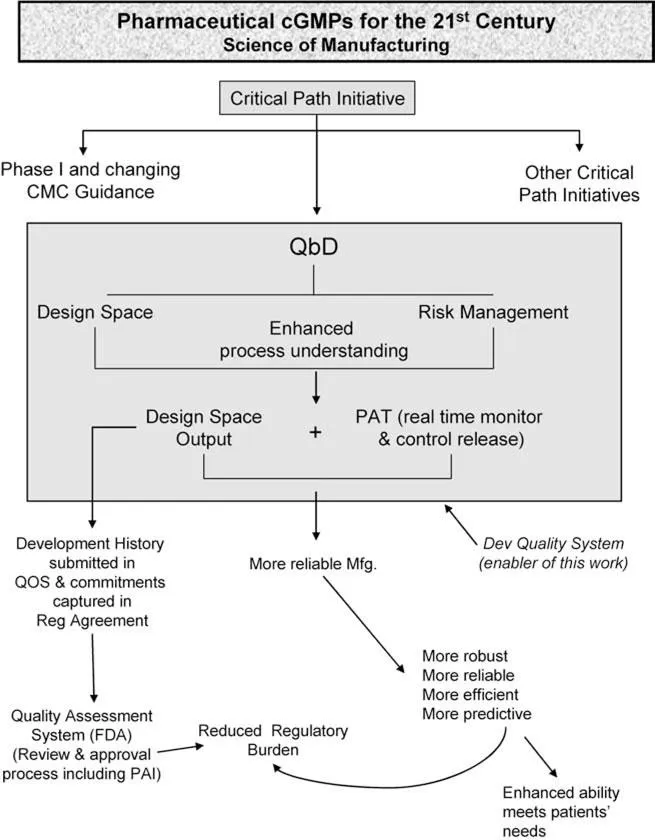
PHARMACEUTICAL CGMPS FOR THE 21ST CENTURY: A RISK-BASED APPROACH
One of the FDA initiatives currently underway that will significantly impact the PAI program is that of “Pharmaceutical cGMPs for the 21st Century: A Risk-Based Approach” (11). This major initiative on the regulation of drug product quality was launched in the...
Table of contents
- Cover
- Half Title
- Title Page
- Copyright Page
- Preface
- Acknowledgements
- Table of Contents
- Contributors
- 1. The Evolution of the Food and Drug Administration: Pre–New Drug Application Approval Inspection
- 2. FDA’s Risk-Based Approach to Inspections
- 4. Training Requirements in Product Development: A Key to a Successful Pre-Approval Inspection
- 5. The Systems-Based Pre-Approval Inspection
- 6. A cGMP Risk Assessment and Management Strategy: Guidelines for the Pre-Approval Inspection
- 7. Concepts in Quality by Design for Drug Development and Manufacture
- 8. Equipment Cleaning During Pharmaceutical Product Development and Its Importance to Pre-Approval Inspection
- 9. Conducting Stability Studies During Development to Ensure Successful Regulatory Approval
- 10. Computer Systems Validation During the Drug Development Process in Anticipation of Pre-Approval Inspections
- 11. Integral for Successful PAI: The Quality Assessment Program
- 12. All Dressed Up but No Approval to Go: The Consequence of Failing an FDA Pre-Approval Inspection
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Preparing for FDA Pre-Approval Inspections by Martin D. Hynes in PDF and/or ePUB format, as well as other popular books in Business & Administrative Law. We have over 1.5 million books available in our catalogue for you to explore.