
eBook - ePub
The Molecular Biology of Neurological Disease
Butterworths International Medical Reviews
- 276 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
The Molecular Biology of Neurological Disease
Butterworths International Medical Reviews
About this book
The Molecular Biology of Neurological Disease reviews advances that have been made in understanding the molecular mechanisms of neurological disorders as well as immediate and future applications of molecular biological techniques to clinical practice. This book explores the molecular genetics of neurological disease such as muscular dystrophy, Joseph disease, and Huntington's disease, along with the mitochondrial genes implicated in such conditions. This text is comprised of 18 chapters and begins by introducing the reader to the basic principles and methods of molecular genetic techniques used in the diagnosis of neurological disease. Attention then turns to several aspects of genetic expression in the brain, including the extent to which the genome is expressed in the brain. The next chapter focuses on the visualization of polyadenylated messenger RNAs in individual cells in mammalian brain using in situ hybridization techniques, combined with immunohistochemical localization of specific proteins and neuropeptides implicated in diseases such as Alzheimer dementia. This book also discusses the molecular biology of chemical synaptic neurotransmission; proteins involved in the regulation of nervous system development; and gene expression in skeletal muscle. This text then concludes with a summary of the ""neurological gene map"" as it stands in the latter part of 1987. This book is intended for physicians who grapple with the problems of neurological disorders on a daily basis, including neurologists, neurologists in training, and those in related fields such as neurosurgery, internal medicine, psychiatry, and rehabilitation medicine.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Topic
MedicineSubtopic
Clinical Medicine1
Molecular genetics and neurological disease: basic principles and methods
A.E. Harding and Roger N. Rosenberg
Publisher Summary
This chapter presents an overview of basic principles and methods related to molecular genetics and neurological disease. Every human cell contains a pair each of 22 chromosomes called the autosomes. In addition, there are either two X chromosomes or one X and one Y chromosome, depending on the sex of the individual. During cell division, the process of mitosis results in two daughter cells which are diploid, that is, each contains an identical chromosomal complement to the parent cell. Gametogenesis involves the more complex process of meiosis, during which each pair of chromosomes exchanges genetic material, resulting in gametes, which are haploid, containing only half the number of chromosomes of the parent cell. Defective genes on the X chromosomes show a distinctive pattern of inheritance in which males are most severely affected and females carrying the gene may be moderately or mildly affected or clinically normal. The variation in expression of an X-linked disorder in females is because of the process of lyonization, during which the expression of one X chromosome is suppressed randomly in each cell.
INTRODUCTION
Prenatal diagnosis of a genetic disease, α-thalassaemia, was first performed using molecular genetic techniques 23 years after the description of the double helical structure of deoxyribonucleic acid (DNA) by Watson and Crick in 1953 (Watson and Crick, 1953; Kan, Golbus and Dozy, 1976). Subsequently, rapid progress has been made in elucidating the molecular basis of inherited disease. At least 3000 disorders exhibiting mendelian inheritance are known to occur in man; 936 of these had been localized to specific chromosomal regions, many using molecular genetic techniques, by 1986 as compared to 300 in 1975 (McKusick, 1986). The molecular genetics of a number of inherited neurological disorders will be discussed in detail in the Chapters 10 to 18. The ‘new genetics’ also has many applications in other neurological and neurobiological problems, including oncology, virology, and development; selected topics pertaining to these are dealt with in Chapters 2–9.
For the clinician or scientist without a strong background in cell biology or genetics, understanding recombinant DNA technology is hampered not only by lack of familiarity with the techniques and their application, but also by the jargon which has been generated by this rapidly expanding field. It is hoped that this chapter will provide a general introduction and glossary to the more specific topics covered by the rest of the book. Many advances in neurogenetics have been made using molecular genetic techniques in genetic linkage studies; this topic will be discussed in some detail, partly for illustrative purposes but also because of its immediate clinical relevance.
PRINCIPLES OF MENDELIAN INHERITANCE
Every human cell contains a pair of each of 22 chromosomes called the autosomes. In addition there are either two X chromosomes or one X and one Y chromosome, depending on the sex of the individual. During cell division the process of mitosis results in two daughter cells which are diploid, that is, each contains an identical chromosomal complement to the parent cell. Gametogenesis involves the more complex process of meiosis, during which each pair of chromosomes exchanges genetic material, resulting in gametes which are haploid, containing only half the number of chromosomes of the parent cell.
Defective genes on the autosomes (chromosomes 1–22) may be inherited as dominant or recessive traits. An individual affected by an autosomal dominant disorder has a 50% chance of transmitting it to offspring (Figure 1.1). In this case, a heterozygous gene carrier manifests the disease despite the presence of a normal corresponding gene (allele) on the other half of the chromosome pair.
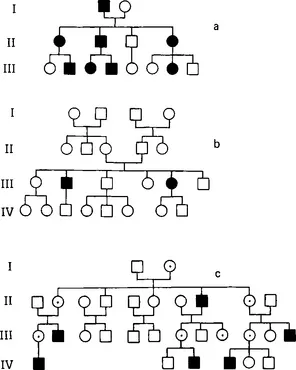
Figure 1.1 Pedigrees illustrating (a) autosomal dominant; (b) autosomal recessive; and (c) X-linked inheritance. Square = male; circle = female; filled symbol = affected; open symbol = unaffected; ⨀ in (c) indicates obligate carrier
This is not so in autosomal recessive inheritance, where it is necessary for both alleles to be abnormal in order for the disease to be expressed (see Figure 1.1), that is, the affected individual is homozygous. We are all heterozygous for one or two recessive genes; the commonest autosomal recessive disorder in the United Kingdom is cystic fibrosis, and about 5% of the population carry the cystic fibrosis gene (Harper, 1981). If two heterozygotes for the same autosomal recessive gene mate, on average one in four of their children will be affected, two out of four will be carriers and one out of four will not carry the gene. It should be obvious that, in populations where family size is small, the majority of individuals with autosomal recessive disorders do not have affected sibs, and often the clinician does not suspect the presence of a genetically determined disorder. Autosomal recessive disorders are more common amongst the offspring of consanguineous parents, for example cousins, as these are more likely to share autosomal recessive genes in common than unrelated members of the population.
Defective genes on the X chromosomes show a distinctive pattern of inheritance in which males are most severely affected, and females carrying the gene may be moderately or mildly affected or clinically normal. The variation in expression of an X-linked disorder in females is due to the process of lyonization, during which the expression of one X chromosome is suppressed randomly in each cell. The distinction between X-linked recessive (in which female carriers are normal) and X-linked dominant (female carriers affected) disorders is rather artificial, although these terms are often used. An important feature of X-linked inheritance is that male-to-male transmission never occurs (see Figure 1.1), but all the female offspring of affected males inherit the abnormal gene.
STRUCTURE AND FUNCTION OF NUCLEIC ACIDS
Both DNA and ribonucleic acid (RNA) consist of sequences of nucleotides, each of which contain a pentose sugar, a phosphate group, and a nitrogenous base. The last may be either pyrimidines (uracil, cytosine, and thymine), or purines (adenine and guanine). DNA contains adenine (A), guanine (G), thymine (T), and cytosine (C) (Figure 1.2). In RNA uracil (U) replaces thymine. RNA is usually single-stranded, but DNA is generally double-stranded, forming an antiparallel double helix; G and C always pair together by means of hydrogen bonds and the same applies to A and T. Compact mammalian chromosomal DNA is supercoiled around proteins called histones. The human genome, that is all the chromosomal DNA, consists of about 3 × 109 base pairs (bp) of DNA.
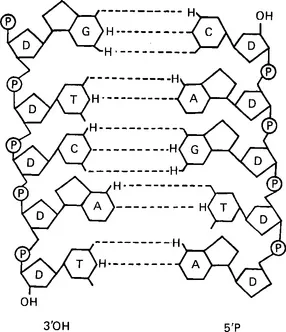
Figure 1.2 The structure of deoxyribonucleic acid. P = phosphate; D = deoxyribose; H = hydrogen; A = adenine; C = cytosine; G = guanine; T = thymine; OH = hydroxyl group. Dotted lines indicate hydrogen bonds between paired nucleotides
Only about 1% of genomic DNA forms structural genes consisting of unique sequences (single copy sequences). Nearly half of human DNA consists of repetitive short sequences which are either not transcribed or, less commonly, encode for small high abundance proteins such as histones. The function of the remaining 50%, some of which codes for introns (non-coding parts of genes which are spliced out after transcription), is also not fully understood. Introns may regulate mRNA stability, thereby increasing the level of the translated gene product. They may exert a major influence on the overall size of mRNA transcripts. For example, the ovalbumin gene, representing some 7000 bases, has only 1859 bases which encode for exons and thus for structural protein. Introns represent the majority of bases in this gene, with approximately 5000 bases present in seven introns (Alberts et al., 1983).
The majority of structural proteins and enzymes appear to be encoded by single copy sequences of DNA. Genes coding for similar functions are sometimes located as compact groups on the same chromosome, such as the immunoglobulin heavy chain gene cluster on chromosome 14. Subunits of multimeric proteins may be encoded by genes scattered throughout the genome, for example the hexosaminidase enzyme complex which is composed of subunits coded for by genes on chromosomes 5 and 15 (McKusick, 1986). Genes may also occur as dispersed families representing multiple copies on several different chromosomes, as has been described for the argininosuccinate synthase and actin genes (Beaudet et al., 1982; McKusick, 1986). It should be stressed that not all of these are transcribed; many may be defective, non-transcribed copies, the so-called pseudogenes. Isogenic copies (different genes coding for the same function) may have tissue specificity. Multiple gene copies can also be expressed on double minute chromosomes, multiple copies of miniature chromosomes which appear to amplify gene action (Schimke et al., 1978; Baskin, Rosenberg and Dev, 1981).
There are also DNA sequences which initiate or stop transcription, the synthesis of messenger RNA (mRNA) complementary to single-stranded DNA sequences catalysed by the enzyme RNA polymerase. This enzyme traverses the DNA template strand from the 3’ to the 5’ end, and synthesizes RNA starting at its 5’ end. The promotor, a specific DNA sequence upstream from the coding region, signals where RNA synthesis is to begin; when RNA polymerase reaches a second DNA sequence, the termination signal, it dissociates from the DNA and releases the RNA chain. A tail of up to 200 adenylic acids may be added to the 3’ end of this chain, and it is capped with methylated guanosine residues at the 5’ end.
In eukaryocytes (higher organisms containing nuclei), nearly all primary RNA trans...
Table of contents
- Cover image
- Title page
- Table of Contents
- Butterworths International Medical Reviews
- Copyright
- Foreword
- Preface
- Contributors
- Chapter 1: Molecular genetics and neurological disease: basic principles and methods
- Chapter 2: Genes expressed in the brain: evolutionary and developmental considerations
- Chapter 3: In situ hybridization: visualizing brain messenger RNA
- Chapter 4: Molecular biology of chemical neurotransmission
- Chapter 5: Proteins which regulate the development of the nervous system
- Chapter 6: Gene expression in skeletal muscle
- Chapter 7: Host and viral genetic factors which influence viral neurotropism
- Chapter 8: Neuro-oncogenesis: recessive genes, activated oncogenes, and chromosome abnormalities in the development of neuroectodermal cancers
- Chapter 9: Transgenic mice and neurological disease
- Chapter 10: Messenger RNA levels in neurological disease
- Chapter 11: Molecular genetics of Joseph disease
- Chapter 12: Huntington’s disease
- Chapter 13: Molecular genetics and muscular dystrophy
- Chapter 14: Mitochondrial genes and neurological disease
- Chapter 15: Molecular basis of retinoblastoma
- Chapter 16: Detection of viral genes in neurological disease
- Chapter 17: Immunogenetics: genetic polymorphism and susceptibility to neurological disease
- Chapter 18: A neurological gene map
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access The Molecular Biology of Neurological Disease by Roger N. Rosenberg,A. E. Harding in PDF and/or ePUB format, as well as other popular books in Medicine & Clinical Medicine. We have over 1.5 million books available in our catalogue for you to explore.