
- 1,024 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
A Comprehensive Guide to Toxicology in Preclinical Drug Development
About this book
A Comprehensive Guide to Toxicology in Preclinical Drug Development is a resource for toxicologists in industry and regulatory settings, as well as directors working in contract resource organizations, who need a thorough understanding of the drug development process. Incorporating real-life case studies and examples, the book is a practical guide that outlines day-to-day activities and experiences in preclinical toxicology. This multi-contributed reference provides a detailed picture of the complex and highly interrelated activities of preclinical toxicology in both small molecules and biologics. The book discusses discovery toxicology and the international guidelines for safety evaluation, and presents traditional and nontraditional toxicology models. Chapters cover development of vaccines, oncology drugs, botanic drugs, monoclonal antibodies, and more, as well as study development and personnel, the role of imaging in preclinical evaluation, and supporting materials for IND applications.
By incorporating the latest research in this area and featuring practical scenarios, this reference is a complete and actionable guide to all aspects of preclinical drug testing.
- Chapters written by world-renowned contributors who are experts in their fields
- Includes the latest research in preclinical drug testing and international guidelines
- Covers preclinical toxicology in small molecules and biologics in one single source
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Topic
MedicineSubtopic
PharmacologyChapter 1
Introduction
Drug development is defined as the entire process of bringing a new drug or device to the market. It involves discovery and synthesis, preclinical development (chemical testing, biological testing, pharmacology, toxicology, safety, etc.), clinical development (Phase I–III), regulatory review, marketing approval, market launch and post-marketing development (Figure 1.1).
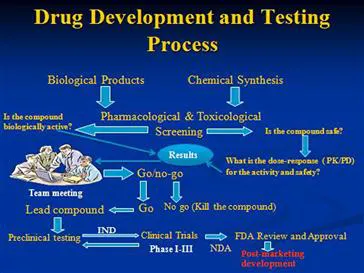
FIGURE 1.1 The drug development process.
The process of drug discovery comprises research on 1) target identification, 2) target prioritization/validation, 3) lead identification, and 4) lead optimization.
A range of techniques are used to identify and isolate individual drug targets. The target identification process isolates drugs that have various interactions with the disease targets and might be beneficial in the treatment of a specific disease. This is followed by a target prioritization phase, during which experimental tests are conducted to confirm that interactions with the drug target are associated with the desired change in the behavior of diseased cells. Identification of lead compounds are sometimes developed as collections, or libraries, of individual molecules that possess the properties required in a new drug. Once the lead is identified, experimental testing is then performed on each of the molecules to confirm their effect on the drug target. This progresses further to lead optimization. Lead optimization studies are conducted on animals or in vitro to compare various lead compounds, to determine how they are metabolized, and what affect they might induce in the body. The information obtained from lead optimization studies helps scientists in the pharmaceutical industry to sort out the compounds with the greatest potential to be developed into a safe and effective drug.
Toxicology studies in the drug discovery process are conducted to evaluate the safety of potential drug candidates. This is accomplished using relevant animal models and validated procedures. The ultimate goal is to translate the animal responses into an understanding of the risk for human subjects. This demands additional studies and investment earlier in the candidate evaluation, coupled with an arduous selection process for drug candidates and a speedy kill to avoid spending money and time on species that would likely fail in development.
Even after a successful drug candidate for a disease target is identified, drug development still faces enormous challenges; which many drugs fail because of their unacceptable toxicity. Safety issues are the leading cause of attrition at all stages of the drug development process and it is important to understand that the majority of safety-related attrition occurs pre-clinically, suggesting that approaches which could identify ’predictable’ preclinical safety liabilities earlier in the drug development process could lead to the design and/or selection of better drug candidates with increased chances of being marketed.
The successful drug candidate undergoes a preclinical safety testing program. Key factors affecting the type of preclinical testing include the chemical structure, nature of the compound (small molecules or biologics), proposed human indication, target population, method of administration, and duration of administration (acute, chronic). During preclinical drug testing, the toxicity and pharmacologic effects of the New Chemical Entity (NCE) are evaluated by in vitro and in vivo laboratory animal testing. Genotoxicity screening is performed, as well as investigations on drug absorption and metabolism, toxicity of the drug’s metabolites, and the speed with which the drug and its metabolites are excreted from the body. Likewise, the drug companies will require a pharmacological profile of the product to be developed, including safety pharmacology – the acute toxicity of the drug in at least two species of animals, and short-term toxicity studies ranging from 2 weeks to 3 months must be conducted, depending on the proposed duration of use of the NCE in the proposed clinical studies. Furthermore, preclinical testing may include chronic toxicity, carcinogenicity, developmental and reproductive toxicology testing. All these studies, together with other specialized study types, such as continuous infusion and photoxicity, are discussed in this book.
It is estimated that it takes eight and more years to develop and test a new drug before it can be approved for clinical use. This estimate includes early laboratory and animal testing, as well as later clinical trials using human subjects.
Preclinical safety data are used to select doses in Phase I clinical trial, to provide information on potential side effects, and thus minimize the risk of serious side effects in clinical trials. It also identifies potential target organs and determines toxicity endpoints not amenable to evaluation in clinical trials such as genetic toxicity, developmental toxicity and carcinogenicity.
Toxicology studies traditionally focus on phenotypic changes in an organism that result from exposure to the drug; therefore, efficient and accurate approaches to assess toxicological effects of drugs on living systems are still less developed. Currently, one of the key factors used for a go/no-go decision making relies on the early knowledge of any potential toxic effect. Thus the traditional approach based on the determination of the No-Observed-Adverse-Effect-Level (NOAEL) is far from accurate. One of the limitations of this approach is that it may fail to detect adverse effects that manifest at low frequencies.
Indeed, in the past 20 years new technologies have emerged that have improved current approaches and are leading to novel predictive approaches for studying disease risk. Increased understanding of the mode of action and the use of scientific tools to predict toxicity is expected to reduce the attrition rate of NCE and thus decrease the cost of developing new drugs. In fact, most big pharmaceutics companies are now using improved model systems for predicting potential drug toxicity, both to decrease the rate of drug-related adverse reactions and to reduce attrition rates. A wide range of biological assay platforms, including toxicogenomics and metabolomics employed in constructing predictive toxicity, are included as separate chapters in this book. The discipline of toxicogenomics is defined as the application of global mRNA, protein and metabolite analysis-related technologies to study the effects of hazards on organisms. Examining the patterns of altered molecular expression caused by specific exposures can reveal how toxicants act and cause their effect. Identification of toxicity pathways and development of targeted assays to systematically assess potential mode of actions allow for a more thorough understanding of safety issues. Indeed, there is high expectation that toxicogenomics in drug development will predict/better assess potential drug toxicity, and hence reduce failure rates.
In addition metabolomics, a more recent discipline related to proteomics and genomics, uses metabolic signatures to determine the molecular mechanisms of drug actions and predict physiological toxicity. The technology involves rapid and high throughput characterization of the small molecule metabolites found in an organism, and is increasingly gaining attention in preclinical safety testing.
This book is a comprehensive guide for toxicologists, regulatory scientists and academics hoping to understand safety testing and the drug development process. It provides a snapshot of the complex and highly interrelated activities of preclinical toxicology in small molecules and biologics. The book also highlights several specific areas, including preclinical drug development of oncogenic and non-oncogenic drugs, oligonucleotides, vaccines, ocular drugs, botanics and monoclonal antibodies. In addition, the book has several unique chapters in areas such as imaging, molecular pathology, abuse liability and biostatistics. The final chapter ‘Practical aspects of developing in-licensed pharmaceutical products’ is intended for small biotech executives with limited funds and resources to advance the drug development process from discovery through to marketing approval. The chapter addresses the chronology of the in-licensing of product candidates.
In closing it must be emphasized that one of the biggest strengths of this book comes from its contributors, who are considered to be authorities in their field. Generally, knowledge of sciences gained through experience in the field shapes personal lives as well as the thinking in the decision making process for day-to-day activities. The experiences of the individual authors currently active in their own specialized areas of interest are carefully crafted in each chapter.
Finally, I would like to thank the contributors for their commitment, and hard work. I also want to express my deep gratitude to Kristine Jones, April Graham, Andy Albrecht and all the production team at Elsevier.
Chapter 2
ADME in Drug Discovery
Outline
Introduction
An Overview of ADME (Absorption, Distribution, Metabolism, Excretion) Science
ADME in Drug Discovery
ADME
Absorption
Physico-Chemical Properties and Permeability
Membrane Bound Drug Transporters
Metabolism in the GIT and Liver: Stability Testing
Distribution and Excretion
In Vivo eADME Disposition and Balance Studies
Drug Distribution Using Molecular Imaging
Metabolism
Biotransformation: Drug Metabolite Profile
Drug-Drug Interactions (DDIs)
Use of Preclinical ADME Data
Two Evolving Technologies Impacting ADME in Drug Discovery
Mass Spectrometry
References
Introduction
An Overview of ADME (Absorption, Distribution, Metabolism, Excretion) Science
The scientific discipline of preclinical drug discovery and development can be described as a risk assessment process, whereby data are used to estimate the usefulness of some agent in preventing, curing, or slowing the progression of human disease. The preclinical phase of research allows clinical studies to be initiated and proceed with some knowledge of risk-benefit. It is an iterative process that varies between different programs at any one time. It is also constantly evolving, as new knowledge and technologies are rapidly introduced. The research plan of today has many general similarities and significant differences from 25 years ago. The constants in this process are drug efficacy and drug safety evaluation, which together represent the Science of Pharmacology, the Science of Drugs. The toxicokinetics, pharmacokinetics in a toxicology study, or the study of the relationship of exposure to toxicity, are important for the design of safety studies (toxicology, safety pharmacology, developmental and reproductive toxicology, etc.). These data allow for estimation (calculation) of a safety margin in preclinical studies and ultimately the early estimation of a Therapeutic Index in humans. In parallel, the study of absorption, distribution, metabolism and excretion are central to finding new, safe and effective drugs. The central message of this chapter is that early characterization of PK (pharmacokinetic) properties is critical to the development of successful drug discovery programs [2–7].
The ADME scientists have two ‘customers’ in the preclinical setting: The drug discovery scientists, who provide new chemical entities for evaluation in various pharmacology and toxicology screens, and the preclinical drug development scientists who provide more refined evaluation of safety and efficacy for preparation of the IND. ADME studies supply the toxicologist with critical measurements of exposure which can be correlated with observed toxicity, which in turn directly relates to Therapeutic Index. Early on in the drug discovery and development process, ADME scientists are interested in estimating clearance (CL), bioavailability (F) and pharmacokinetic/pharmacodynamic (PK/PD) data for entry into compound libraries. In addition, ADME scientists are charged with providing to their toxicology colleagues an understanding of exposure and toxicity, the PK/PD (or TK/TD; toxicokinetic/toxicodynamic) relationship and an assessment of the role of metabolism, transporters, drug metabolizing enzymes and drug accumulation in drug safety. This chapter will address ADME in discovery research, or ADME at the interface of drug discovery and drug development, which is commonly now referred to as early-ADME (eADME). Not all topics will be covered. For example, plasma protein binding (PPB) has been omitted, since it is less important than critical concepts such as stability and clearance [8].
The characterization of ADME properties of compounds early in the drug discovery process has well characterized value for the selection of better drug candidates, and has become more important as technologies impacting this process have developed and matured [9–11]. The cytochrome P450 (CYPs) enzymes are intimately involved in ADME. The catalytic cycle of the P450-dependent monooxygenase system is displayed in Figure 2.1 (showing the second electron insertion step from cytochrome b5). Over the last 20 years, an understanding of the biochemistry of the Cytochrome P-450 system and the role that CYP inhibition, CYP phenotype and CYP induction plays in the identification of better drug therapies has impacted how preclinical ADME research is conducted [12–14]. Consider that 20 years ago approximately 40% of clinical drug failures could be tied to PK and ADME problems, and today this failure rate is 10% or less for companies with comprehensive, state-of-the-art preclinical discovery/development programs addressing these issues [15]. The drug discovery process continues to evolve and early ADME evaluation has become a routine part of the ‘Big Picture’ process to examine the utility of drug templates in the discovery of novel therapeutics. At time of writing, the FDA released Guidance for Industry, Drug Interaction Studies, Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations, which provide much needed regulatory guidance for many of the ADME investigations discus...
Table of contents
- Cover image
- Title page
- Table of Contents
- Copyright
- Dedication
- Foreword
- Contributors
- Chapter 1. Introduction
- Chapter 2. ADME in Drug Discovery
- Chapter 3. Pharmacokinetics and Toxicokinetics
- Chapter 4. Development of Preclinical Formulations for Toxicology Studies
- Chapter 5. Acute, Sub-Acute, Sub-Chronic and Chronic General Toxicity Testing for Preclinical Drug Development
- Chapter 6. Contemporary Practices in Core Safety Pharmacology Assessments
- Chapter 7. Genetic Toxicology Testing
- Chapter 8. Clinical Pathology
- Chapter 9. Best Practice in Toxicological Pathology
- Chapter 10. Molecular Pathology: Applications in Nonclinical Drug Development
- Chapter 11. Infusion Toxicology and Techniques
- Chapter 12. The Preparation of a Preclinical Dossier to Support an Investigational New Drug (IND) Application and First-in-Human Clinical Trial
- Chapter 13. Developmental and Reproductive Toxicology
- Chapter 14. Immunotoxicology Assessment in Drug Development
- Chapter 15. Juvenile Toxicity Testing to Support Clinical Trials in the Pediatric Population
- Chapter 16. Photosafety: Current Methods and Future Direction
- Chapter 17. Preclinical Evaluation of Carcinogenicity using the Rodent Two-Year Bioassay
- Chapter 18. Carcinogenicity Evaluations using Genetically Engineered Animals
- Chapter 19. Current Strategies for Abuse Liability Assessment of New Chemical Entities
- Chapter 20. Impact of Product Attributes on Preclinical Safety Evaluation
- Chapter 21. Preclinical Development of Monoclonal Antibodies
- Chapter 22. Preclinical Development of Non-Oncogenic Drugs (Small and Large Molecules)
- Chapter 23. Preclinical Development of Oncology Drugs
- Chapter 24. Safety Evaluation of Ocular Drugs
- Chapter 25. Preclinical Toxicology of Vaccines
- Chapter 26. Overview of the Nonclinical Development Strategies and Class-Effects of Oligonucleotide-Based Therapeutics
- Chapter 27. Nonclinical Safety Assessment of Botanical Products
- Chapter 28. Regulatory Toxicology
- Chapter 29. New Drug Regulation and Approval in China
- Chapter 30. Biostatistics for Toxicologists
- Chapter 31. Role of Study Director and Study Monitor in Drug Development
- Chapter 32. Use of Imaging for Preclinical Evaluation
- Chapter 33. Predictive Toxicology: Biological Assay Platforms
- Chapter 34. Toxicometabolomics: Technology and Applications
- Chapter 35. Toxicogenomics in Preclinical Development
- Chapter 36. Practical Aspects of Developing In-Licensed Pharmaceutical Products: The Virtual Development Paradigm
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access A Comprehensive Guide to Toxicology in Preclinical Drug Development by Ali S. Faqi in PDF and/or ePUB format, as well as other popular books in Medicine & Pharmacology. We have over 1.5 million books available in our catalogue for you to explore.