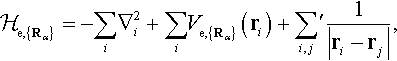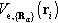Multiscale materials modelling offers an integrated approach to modelling material behaviour across a range of scales from the electronic, atomic and microstructural up to the component level. As a result, it provides valuable new insights into complex structures and their properties, opening the way to develop new, multi-functional materials together with improved process and product designs. Multiscale materials modelling summarises some of the key techniques and their applications.The various chapters cover the spectrum of scales in modelling methodologies, including electronic structure calculations, mesoscale and continuum modelling. The book covers such themes as dislocation behaviour and plasticity as well as the modelling of structural materials such as metals, polymers and ceramics. With its distinguished editor and international team of contributors, Multiscale materials modelling is a valuable reference for both the modelling community and those in industry wanting to know more about how multiscale materials modelling can help optimise product and process design.
- Reviews the principles and applications of mult-scale materials modelling
- Covers themes such as dislocation behaviour and plasticity and the modelling of structural materials
- Examines the spectrum of scales in modelling methodologies, including electronic structure calculations, mesoscale and continuum modelling
Trusted by 375,005 students
Access to over 1 million titles for a fair monthly price.
The role of ab initio electronic structure calculations in multiscale modelling of materials
M. Šob Masaryk University, Czech Republic and Academy of Sciences of the Czech Republic
1.1 Introduction
Most, if not all, of the properties of solids can be traced to the behavior of electrons, the ‘glue’ that holds atoms together to form a solid. An important aim of the condensed matter theory is thus calculating the electronic structure (ES) of solids. The theory of ES is not ony helpful in understanding and interpreting experiments, but it also becomes a predictive tool of the physics and chemistry of condensed matter and materials science.
Many of the structural and dynamical properties of solids can be predicted accurately from ab initio (first-principles) electronic structure calculations, i.e. from the fundamental quantum theory. Here the atomic numbers of constituent atoms and some structural information are employed as the only pieces of input data. Such calculations are routinely performed within the framework of density functional theory in which the complicated many-body motion of all electrons is replaced by an equivalent but simpler problem of a single electron moving in an effective potential.
A general formulation of the quantum mechanical equations for ES including all known interactions between the electrons and atomic nuclei in solids is relatively simple, but we are still not able to solve these equations in their full generality. A great many approximations must be performed, which, in many cases, leads to a comprehensive solution. Its analysis brings us then some understanding of various phenomena and processes in condensed matter. The ES problem is computationally very demanding. This is why practical ES calculations in solids were rather rare prior to the availability of larger high-speed computers.
Since the 1980s, ES theory has exhibited a growing ability to understand and predict material properties and to use computers to design new materials. A new field of solid-state physics and materials science has emerged – computational materials science. This has achieved a considerable degree of reliability concerning predictions of physical and chemical properties and phenomena, thanks in large part to continued rapid development and availability of computing power (speed and memory), its increasing accessibility (via networks and workstations), and to the generation of new computational methods and algorithms which this enabled. State-of-the-art ES calculations yield highly precise solutions to the one-electron Kohn–Sham equation for a solid and provide an understanding of matter at the atomic and electronic scale with an unprecedented level of detail and accuracy. In many cases, we are able not only to simulate experiment but also to design new molecules and materials and to predict their properties before actually synthesizing them. A computational simulation can also provide data on the atomic scale that are inaccessible experimentally. In contrast to semi-empirical approaches – the adjustable parameters of which are fitted to the properties of the ground state structure and, therefore, may not be transferable to non-equilibrium configurations – ab initio calculations are reliable far from the equilibrium as well.
In multiscale modelling of materials, the role of ab initio electronic structure calculations is twofold: (i) to study the situations where the electronic effects are crucial and must be treated from first principles and (ii) to provide data for generation of interatomic potentials. In this chapter, we will discuss both these aspects. Let us note that there exists a vast literature devoted to multiscale modelling of materials. Recent reviews may be found for example in1,2 and in the Handbook of Materials Modeling edited by S. Yip3, the latest developments are documented in the proceedings of various meetings on multiscale modelling of materials (the latest one took place in September 2006 in Freiburg, Germany4).
1.2 Basic equations of electronic structure calculations
In a solid where relativistic effects are not essential, we may describe the electron states by the non-relativistic many-electron Schrödinger equation
[1.1]
with the Hamiltonian
[1.2]
where {Rα} are the instantaneous positions of the atomic nuclei, {ri} denote positions of electrons, the
is the potential experienced by the ith electron in the field of all nuclei at the positions {Rα} with the atomic numbers Zα, i.e.
[1.3]
and the last term in equation [1.2] represents the electrostatic electron–electron interaction (the prime on the summation excludes i = j). Let us note that here and throughout the chapter we use Rydberg atomic units with ħ = 1, 2me = 1 and e2 = 2, where me and e denote the electron mass and charge, ...
Table of contents
Cover image
Title page
Table of Contents
Copyright page
Contributor contact details
Preface
1: The role of ab initio electronic structure calculations in multiscale modelling of materials
2: Modelling of dislocation behaviour at the continuum level
3: Phase-field modelling of material microstructure
4: Mesoscale modelling of grain growth and microstructure in polycrystalline materials
5: Finite element and homogenization modelling of materials
6: Grain–continuum modelling of material behaviour
7: Coupled atomistic/continuum modelling of plasticity in materials
8: Multiscale modelling of carbon nanostructures
9: Multiscale modelling of structural materials
Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.4M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
Both plans are available with monthly, semester, or annual billing cycles.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go. Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Multiscale Materials Modelling by Z. X. Guo in PDF and/or ePUB format, as well as other popular books in Technology & Engineering & Materials Science. We have over one million books available in our catalogue for you to explore.