
eBook - ePub
Specification of Drug Substances and Products
Development and Validation of Analytical Methods
- 390 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Specification of Drug Substances and Products
Development and Validation of Analytical Methods
About this book
Specification of Drug Substances and Products: Development and Validation of Analytical Methods is a comprehensive and critical analysis of the requirements and approaches to setting specifications for new pharmaceutical products, with an emphasis on phase-appropriate development and validation of analytical methods. This book is intended as more than a review of new regional guidelines, existing regulatory guidance, and industry practices. It provides a hands-on guide to understanding and applying these in practice. The authors discuss critical issues, novel approaches, and future directions while also providing insight into how International Guidelines were developed and the rationale behind them.
- Guide to industry best practices of analytical methodologies used in the specification of new drug substances and products (e.g. DOE, QbD)
- Critical assessment of the application of ICH guidelines on method validation and specification setting, written by experts involved in the development and application of the guidelines to aid understanding of requirements and what is expected by regulatory authorities
- Direct applicability to the day-to-day activities in drug development and the potential to increase productivity
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Topic
Physical SciencesSubtopic
Analytic ChemistryPart 1
Introduction
Outline
Chapter 1 Introduction
Chapter 2 General principles and regulatory considerations
Chapter 3 Application of quality by design (QbD) to the development and validation of analytical methods
Chapter 4 General principles and regulatory considerations
Chapter 1
Introduction
Christopher M. Riley∗, Bradford J. Mueller†, Thomas W. Rosanske∗∗ and Shelley R. Rabel Riley∗, ‡, ∗Riley and Rabel Consulting Services, Maryville, MO, USA, †Incyte Corporation, Experimental Station, Wilmington, DE, USA, ∗∗T.W. Rosanske Consulting, Overland Park, KS, USA, ‡Department of Natural Sciences, Northwest Missouri State University, Maryville, MO, USA
Chapter Outline
References
When the first version of this book was published in 1996,1 The International Conference on Harmonization (ICH), which is an effort by the USA, the EU and Japan to harmonize new drug applications, was still in its infancy. Since then, all the key ICH Quality Guidelines2–26 covering specification setting (e.g. ICH Q1,2–8 Q3–Q6)9–21 and method validation (ICH Q2)6 have been published, and some have been revised at least once. The ICH Guidelines, together with some of the more recent changes in regional guidelines and compendial requirements will form the general framework for this book. Where the Quality (Q1–Q11) ICH Guidelines fit into the general drug development framework is shown in Fig. 1.1.
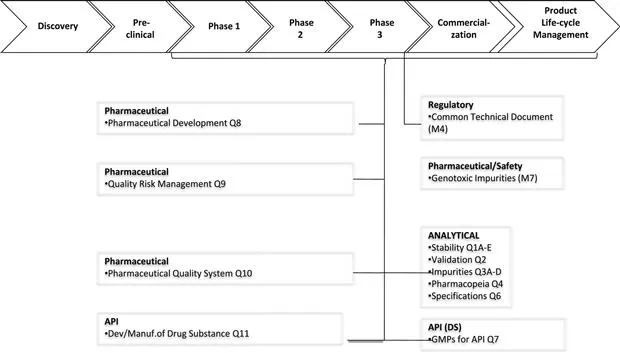
FIGURE 1.1 Summary of the ICH Guidelines applicable to pharmaceutical analysis (see also Refs 2–25) and where they fit into the drug development process.
The introduction of the earlier ICH Quality Guidelines (Q1–Q6),2–21 which describe most of the general requirements for the analytical content of the Common Technical Document (CTD, ICH M4Q(R1))22 and its electronic counterpart (eCTD), was followed by a series of guidelines (Q7–Q10) addressing some of the key approaches to drug development that are also to be included in the CTD. Although there are some regional differences, the CTD is the generally harmonized document used in the ICH regions for marketing authorization applications. The general framework of the CTD is also used, with appropriate modifications, for clinical trials applications. The CTD is also accepted in many non-ICH countries, such as Canada and Australia.
According to the ICH definition, the specification(s) for a new drug substance or a drug product (Q6A and Q6B) contain three elements: (1) the quality attributes (or tests), (2) references to the associated methods and (3) the acceptance criteria. The primary objective of this book is to provide a critical and comprehensive assessment of the approaches used to identify what are the key quality attributes that impact safety, efficacy, and manufacturability, select appropriate analytical methods based on the accuracy and precision needed to adequately measure and control the identified quality attributes and determine how the analytical methods are developed and validated for their intended use. The general principles of the specification-setting process are surveyed in Chapter 2 and explored in greater detail in Chapters 5–15. Chapter 16 deals with the development and validation of bioanalytical methods.
The concept of Quality by Design (QbD) was introduced into the drug development process through the more recent ICH Guidelines (Q8–Q10)23–25 with the primary aim of increasing the understanding and the knowledge base of the processes for the manufacturing of drug substances and products. However, the principles of QbD are equally applicable to pharmaceutical analysis. Therefore, the concept of Analytical Quality by Design (AQbD) is introduced in Chapter 3 and expanded in later chapters. Since the publication of the first version of this book, the key ICH Quality Guidelines have matured and now form the general framework for the application of worldwide marketing approvals of new drug products.
Whereas the guidelines dealing with specification setting (most notably ICH Q6A and Q6B) and Method Validation (Q2) describe what information regulators expect to see in a new drug application, they provide very little detail on how the guidelines are to be implemented at the technical level. The absence of specific direction on the implementation of the ICH Quality Guidelines allows for the application of new and improved analytical technologies targeted to the critical quality attributes which impact product performance. The use of statistical approaches to better correlate method performance with respect to control limits for critical quality attributes and to monitor long-term analytical method performance is an area which is not discussed within the guidances, but is critical to the development and maintenance of analytical methods. Whereas Chapters 2 and 3 survey the general principles of specification setting and QbD, respectively, Chapter 4 discusses conventional approaches to method validation. The ICH Guideline on Method Validation (Q2(R1)) was primarily developed with separation techniques in mind and the following tests in particular:
• Identification tests
• Quantitative tests for impurities content
• Limit tests for the control of impurities
• Quantitative tests of the active moiety in samples of drug substance and drug product or other selected components in the drug product (e.g. preservatives, antioxidants)
Subsequent chapters will discuss how the principles of method validation set forth in Q2(R1) have been adapted to techniques as diverse as solid characterization and microbiological methods.
In keeping with the spirit of the first version of this book, this version is not intended as merely a review of existing regulatory guidance and industry practices. Rather, in addition to discussing conventional approaches, each chapter will address critical issues and novel approaches. The authors have been carefully selected as being former members of the ICH Expert Working Groups charged with developing the ICH guidelines, and/or subject-matter experts in the industry, academia and government laboratories. Thus, the book will provide the reader with not only an understanding of industry best practices and future directions, but also an insight into how international guidelines were developed and the rationale behind them.
In addition to providing the “what” but not the “how” to set specifications and validate analytical methods, the ICH Quality Guidelines (Q1–Q6)2–20 only define what is to be provided in a new drug application. They expressly exclude what is expected in the clinical stages of drug development (i.e. in an Investigational New Drug Application, IND). Therefore, a common theme throughout the book is how the methods are validated and specifications evolve over the drug development life cycle (Fig. 1.2). The intention in writing the second version of the book is to capture the many regulatory and technical advances that have occurred in the field since publication of the first version in 1996.
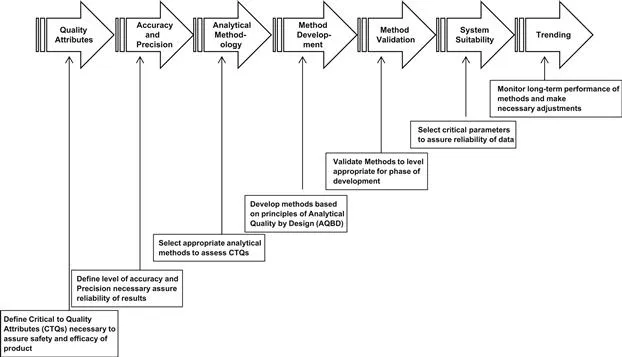
FIGURE 1.2 The evolution of analytical technology for the control of pharmaceuticals through the life cycle of the development process.
The “how” of the earlier Q1–Q6 Guidelines are to be applied is described in large part in subsequent guidelines (Q7–Q11). For example,27 16 attributes were identified for a polymeric excipient, derived from a natural product, and used in sustained release product to control the potentially variable performance of the excipient in the product. The only way to manage the 16 attributes and achieve acceptable product performance was to understand the contributions of the various attributes and the interactions between them—between each physical and chemical characteristic. By analytically measuring each of the attributes and then using statistical/chemometric approaches, it was possible to define a “design space” of all parameters which could deliver the overall desired effect of drug release.
Thus, this version is intended to be not only a review of the ICH Guidelines relating to the specification and method validation of new drugs, but also to provide a critical analysis of the regulatory guidelines and a comprehensive treatment of how those guidelines are applied to the development of new drugs. It is intended to be an educational tool and a reference source for those involved in the development and regulation of new drug products.
References
1. Development and Validation of Analytical Methods. In: Riley CM, Rosanske TW, eds. Elsevier 1996; Progress in Pharmaceutical and Biomedical Analysis. Vol. 3.
2. Stability Testing of New Substances and Products (Q1A(R2)). The International Conference on Harmonization of Technical Requirements for Registration of...
Table of contents
- Cover image
- Title page
- Table of Contents
- Copyright
- List of Contributors
- Part 1: Introduction
- Part 2: Universal Tests
- Part 3: Specific Tests: Drug Substance
- Part 4: Specific Tests: Drug Product
- Part 5: Pharmacopeial Methods
- Part 6: Microbial Methods
- Part 7: Biological Fluids
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Specification of Drug Substances and Products by Christopher M. Riley, Thomas W. Rosanske, Christopher M. Riley,Thomas W. Rosanske in PDF and/or ePUB format, as well as other popular books in Physical Sciences & Analytic Chemistry. We have over 1.5 million books available in our catalogue for you to explore.