
eBook - ePub
Magnetic Resonance Spectroscopy
Tools for Neuroscience Research and Emerging Clinical Applications
- 398 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Magnetic Resonance Spectroscopy
Tools for Neuroscience Research and Emerging Clinical Applications
About this book
Magnetic Resonance Spectroscopy: Tools for Neuroscience Research and Emerging Clinical Applications is the first comprehensive book for non-physicists that addresses the emerging and exciting technique of magnetic resonance spectroscopy. Divided into three sections, this book provides coverage of the key areas of concern for researchers. The first, on how MRS is acquired, provides a comprehensive overview of the techniques, analysis, and pitfalls encountered in MRS; the second, on what can be seen by MRS, provides essential background physiology and biochemistry on the major metabolites studied; the final sections, on why MRS is used, constitutes a detailed guide to the major clinical and scientific uses of MRS, the current state of teh art, and recent innovations.
Magnetic Resonance Spectroscopy will become the essential guide for people new to the technique and give those more familiar with MRS a new perspective.
- Chapters written by world-leading experts in the field
- Fully illustrated
- Covers both proton and non-proton MRS
- Includes the background to novel MRS imaging approaches
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
Section 1
Technical Aspects—How MRS is Acquired
Outline
Chapter 1.1 Basis of Magnetic Resonance
Chapter 1.2 Localized Single-Voxel Magnetic Resonance Spectroscopy, Water Suppression, and Novel Approaches for Ultrashort Echo-Time Measurements
Chapter 1.3 Technical Considerations for Multivoxel Approaches and Magnetic Resonance Spectroscopic Imaging
Chapter 1.4 Spectral Editing and 2D NMR
Chapter 1.5 Spectral Quantification and Pitfalls in Interpreting Magnetic Resonance Spectroscopic Data
Chapter 1.1
Basis of Magnetic Resonance
Christoph Juchem1,2 and Douglas L. Rothman1, 1Department of Diagnostic Radiology, 2Department of Neurology, Yale School of Medicine, Massachussetts, USA
Magnetic resonance spectroscopy (MRS) allows the detection and quantification of chemical compounds from localized portions of the living tissue, e.g., the brain, in a noninvasive fashion. The goal of this chapter is to provide an introduction to and summarize commonly applied in vivo MRS methods. The descriptions in this chapter will be kept at a relatively introductory level focusing on the basic MR physics and engineering principles involved. More information on the experimental implementation and applications of these methods are provided in subsequent chapters of this book.
Keywords
MR physics; MRS; quantitation; spectroscopy
Introduction
Magnetic resonance spectroscopy (MRS) allows the detection and quantification of chemical compounds from localized portions of the living tissue, e.g., the brain, in a noninvasive fashion. It thereby provides a powerful tool to assess key aspects of brain metabolism and function. In the clinics, the repertoire of measurable compounds along with the quantitative character of the derived information makes MRS a versatile tool for the identification of clinical conditions, for longitudinal patient monitoring and for treatment control. The goal of this chapter is to summarize the basic concepts of commonly applied in vivo MRS methods. These concepts will be developed more in subsequent chapters as well as more advanced MRS methods such as spectroscopic imaging and J-editing, and measurement of specific chemicals and metabolic pathways. The descriptions in this chapter will be kept at a relatively basic level. For a more advanced but still very accessible treatment of basic MRS spin physics as well as MRS methodology we recommend de Graaf (2008).
In Vivo Spectroscopy
Atomic nuclei with a magnetic moment (or “spin”) can exhibit resonance behavior in a magnetic field. This magnetic resonance effect is governed by the Larmor equation as a simple linear relationship between the magnetic field B perceived by the nucleus and the resultant resonance frequency ω.
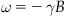
The relative scaling γ, the so-called gyromagnetic ratio, is a nucleus-specific constant. The hydrogen nucleus 1H (i.e., a proton) is the prime example for an MR-sensitive isotope as 1H resonance signals from hydrogen nuclei bound in tissue water provide the basis for MR imaging (MRI) of the human body. Nuclear spins can be thought of as microscopic magnets. When placed in a magnetic field these magnetic moments become polarized, and are either parallel or antiparallel to the field. The spins that are polarized parallel to the field are in a lower energy state, so slightly more are present in this alignment than antiparallel to the field. When radiofrequency (RF) energy is applied to spins in a magnet at the Larmor frequency, the spins will absorb energy and undergo a transition from the antiparallel to the parallel state as with optical and other forms of resonance spectroscopy. MRS, however, differs in that in the process of absorption the spins will become polarized with the RF field such that when the RF field is shut off the spins will effectively rotate along the axis of the magnet (by convention the z-axis in MRS). This phenomenon creates a rotating magnetic field at the Larmor frequency. When an RF receiver coil is used the rotating magnetization induces an oscillating voltage by induction, which is then detected by the MR spectrometer (which is essentially a large RF transmitter and receiver). At field strengths available for clinical MRS the Larmor frequency will vary from ~60 to 300 MHz corresponding to magnet field strengths of 1.5 and 7.0 T, respectively. The low resonance frequency (6 to 7 orders of magnitude lower than optical resonances) is disadvantageous from a sensitivity standpoint, because the energy per absorption or emission is proportional to frequency. However, unlike optical spectroscopy or imaging, the human body is relatively transparent to RF at the MHz range and therefore is completely accessible to measurement by MRS.
When a human subject is placed in the scanner of a given field strength, one could assume that all of the body’s 1H nuclei exhibit the same resonance frequency. In reality, however, minute frequency variations are observed depending on the molecular structure the atomic nucleus is embedded in. The field variations to cause these frequency shifts are based on two different effects. The electronic, i.e., the chemical, environment around the atomic nucleus at hand results in a so-called “chemical shift” and the correlation of different nuclei of the same molecule mediated through their binding electrons is referred to as dipolar or J-coupling. Chemical shift and J-coupling critically rely on the molecules’ chemical and geometric composition. As such, their measurement provides a wealth of intramolecular, microscopic information from a relatively simple, “macroscopic” MRS experiment. Notably, chemical shift and J-coupling are the basis for the key role MRS is playing in structural and analytical chemistry. Although chemical shift and J-coupling were discovered in the 1950s, it was not until 20 years later that MRS was applied to identify and quantify biochemicals in living cells (Shulman et al., 1979) and eventually in vivo (Ackerman et al., 1980). With this paradigm shift, the goal was no longer to study the physicochemical properties of substances, but to use the knowledge on the substance-specific spectroscopic patterns, i.e., their spectroscopic fingerprint, to separate and quantify these substances in vivo to infer the concentrations of metabolites and the fluxes of metabolic pathways.
MRS allows the noninvasive quantification of neurochemicals that contain MR-sensitive isotopes (e.g., 1H). Hydrogen is prevalent in most metabolites of the human brain and the 1H nucleus is the most relevant isotope for in vivo MRS. The gyromagnetic ratio of 1H is highest for all stable isotopes; therefore, its sensitivity in MR experiments is higher than for any other nucleus. Furthermore, the natural abundance of the 1H nucleus is almost 100%. The first in vivo MRS measurements of the living brain were in 1982 on a small-bore high-field MR system (Behar et al., 1983). With the subsequent rapid development of large-bore high-field magnets and volume localization by the mid-1980s, the first 1H MR spectra of human brain were obtained (Bottomley et al., 1983; Frahm et al., 1989) and today 1H MR spectra can be somewhat routinely obtained on all clinical MRI systems of 1.5 T and higher in field strength.
Although a series of brain metabolites can be identified with 1H MRS (Govindaraju et al., 2000), the number of substances that are assessable under in vivo conditions does not exceed 15–20 (Mekle et al., 2009; Tkac et al., 2009; Emir et al., 2011a) and is typically well below. Further MR-visible isotopes with biochemical relevance have been shown to provide valuable information on tissue physiology and biochemistry. Phosphorus (31P) MRS, for instance, allows the quantification of key components of the tissues’ energy metabolism (ATP, ADP, and phosphocreatine) and to study, for example, bioenergetic deficits in response to disease (Befroy & Shulman, 2011). MRS employing the 13C isotope enables the study of Krebs (TCA) cycle intermediates, and the predictable transfer of infused 13C-label between specific positions of the carbon chains of these intermediates stands out by providing turnover rates and metabolic fluxes (Rothman et al., 2011). Today’s clinical MRS, however, mostly relies on 1H whereas other nuclei are predominantly used in basic and preclinical research. The need for nonstandard hardware and specialized MRS methods for non-1H MRS might be reasons why these applications have yet to prevail in clinical practice. This chapter therefore focuses on the basic principles of 1H MRS, although where appropriate extensions to other nuclei will be discussed (for more detail on this topic see Chapters 4.1–4.3.).
MRS Methods
Basics of MRS
The goal of in vivo 1H MRS is to quantify biochemical substances in a noninvasive fashion. In general, the signal amplitudes of an MRS acquisition and the resultant peaks in the reconstructed spectrum directly scale with the number of resonating nuclei and the substance amount seen by the MRS experiment. As such, spectral peaks and patterns provide a direct measure for the concentration of the metabolite at hand. Water is by far the most prevalent 1H-containing compound in human tissue; therefore it contributes the largest part of the MR signal (Fig. 1.1.1A). Since the water concentration itself is of little clinical relevance, its signal is typically minimized in MRS experiments to better reveal the metabolites of interest (Fig. 1.1.1B). A prominent limitation of 1H MRS of brain metabolites is the small frequency spread between spectral peaks. This limited spectral dispersion leads to severe spectral overlap of the observed patterns and poses a significant challenge for the identification and separation of the individual compounds. Notably, the problem of spectral overlap is further complicated in vivo by limited magnetic field homogeneity and the resultant broadening of spectral lines. The strongest limitation for the quantification of tissue metabolites, however, arises from the inherently poor sensitivity of MR methods. Metabolite concentrations in the millimolar range are therefore necessary with 1H MRS to achieve signal amplitudes that sufficiently exceed the inevitable measurement noise. Moreover, adequate signal-to-noise ratio (SNR) is particularly relevant when partially overlapping spectral structures are to be disentangled, as is typically the case for 1H MRS.
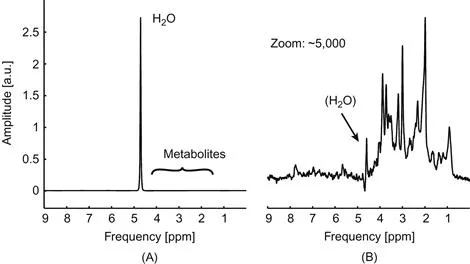
Figure 1.1.1 The role of water suppression for in vivo 1H MRS of the human brain. (A) The water content in human brain tissue largely exceeds the concentrations of other observable substances and therefore dominates the appearance of the spectrum. (B) Suppression of the water signal allows an unobstructed vie...
Table of contents
- Cover image
- Title page
- Table of Contents
- Copyright
- Acknowledgements
- Contributors
- Introduction
- Section 1: Technical Aspects—How MRS is Acquired
- Section 2: Biochemistry — What Underlies the Signal?
- Section 3: Applications of Proton-MRS
- Section 4: Applications of Non-Proton MRS
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Magnetic Resonance Spectroscopy by Charlotte Stagg,Douglas L. Rothman in PDF and/or ePUB format, as well as other popular books in Medicine & Neurology. We have over 1.5 million books available in our catalogue for you to explore.