
- 284 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Muscular Dystrophies
About this book
The Handbook of Clinical Neurology Vol 101: Muscular Dystrophies discusses the pathogenesis and treatment prospects for muscular dystrophies. It summarizes the advances in molecular and cell biology, biochemistry, and other biological sciences, with an emphasis on their application to this group of muscle disorders and to their clinical implications.
Starting with an overview of muscular dystrophies, the book's 16 chapters discuss dystrophinopathies; sarcoglycanopathies; congenital muscular dystrophies; collagen VI-related myopathies; limb-girdle muscular dystrophy 2A; dysferlinopathies; limb-girdle muscular dystrophy 2H and the role of TRIM32; and caveolinopathies. The book also covers myofibrillar myopathies; Emery–Dreifuss muscular dystrophy; facioscapulohumeral dystrophy and scapuloperoneal syndromes; oculopharyngeal muscular dystrophy; myotonic dystrophy types 1 and 2; and distal muscular dystrophies.
This book is useful to basic investigators, as it offers an increased understanding of muscular dystrophies; and to clinicians, with its emphasis on issues that are relevant to the care, diagnosis, and management of patients with these disorders.
- Valuable insights into the muscular dystrophies, including treatment, diagnosis, and care and patient management
- A comprehensive compilation of the combined wisdom of the most highly regarded physicians, experts, and scientists studying the muscular dystrophies
- An evaluation of the way advances in molecular and cell biology, biochemistry, and other biological sciences continue to advance the study of these disorders
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Handbook of Clinical Neurology, Vol. 101, No. Suppl C, 2011
ISSN: 0072-9752
doi: 10.1016/B978-0-08-045031-5.00001-3
Chapter 1Overview of the muscular dystrophies
Abstract
The muscular dystrophies are a clinically and genetically heterogeneous group of myopathies typically associated with progressive weakness. Weakness may be noted at birth or develop in late adult life. Some patients manifest with myalgias, rhabdomyolysis, or only raised serum creatine kinase levels without any symptoms or signs of weakness. The muscular dystrophies can be inherited in an X-linked, autosomal recessive, or autosomal dominant fashion and can result from mutations affecting structural proteins localizable to the sarcolemmal proteins, nuclear membrane, basement membrane, sarcomere, or nonstructural enzymatic proteins. This chapter provided a brief overview of the muscular dystrophies before later chapters discuss the individual subtypes in greater detail.
Introduction
Classification
Historically, the muscular dystrophies have been defined as progressive myopathies in which muscle biopsies demonstrate replacement of muscle fibers by adipose and connective tissue. The clinical onset of the dystrophy may be evident at birth, as in congenital muscular dystrophies, or may not develop until late adulthood. The dystrophies were felt to differ from congenital myopathies by the presence of specific ultrastructural abnormalities apparent in muscle biopsies in the latter (e.g., nemaline rods, cores, or minicores). However, with advances in molecular genetics the distinction between what constitutes a muscular dystrophy and a congenital myopathy has become blurred. For example, myofibrillar abnormalities and inclusions such as nemaline rods, cytoplasmic bodies, and reducing bodies that initially led to categorization as a congenital myopathy have been noted in disorders known to carry similar genetic defects that have been considered forms of dystrophy.
Dystrophies have been classified according to age of onset, mode of inheritance, and pattern of weakness (Table 1.1). For example, those that present at birth have been termed congenital muscular dystrophies (MDC). Dystrophies have also been named based on the patterns of muscle involvement, including limb-girdle muscular dystrophy (LGMD), facioscapulohumeral dystrophy (FSHD), oculopharyngeal muscular dystrophy (OPMD), distal myopathy/dystrophies, and scapuloperoneal dystrophy. Within the distal muscular dystrophies, subclassifications have been based on inheritance pattern, age of onset, and the specific muscle groups initially affected; for example, the Markesbery–Griggs, Udd, and Laing types of distal myopathy have preferential involvement of the anterior tibial muscles, Miyoshi myopathy the gastrocnemius, and Welander myopathy the extensor forearm muscles. Dystrophies associated with proximal greater than distal weakness are called limb-girdle dystrophies (LGMD). The LGMD, inherited in an autosomal dominant fashion, are termed LGMD type 1 (LGMD1), whereas autosomal recessive dystrophies are called LGMD2. Further subclassifications of the LGMDs are based on genotype differences (e.g., LGMD1A, LGMD1B).
Table 1.1 Genetic classification of the muscular dystrophies
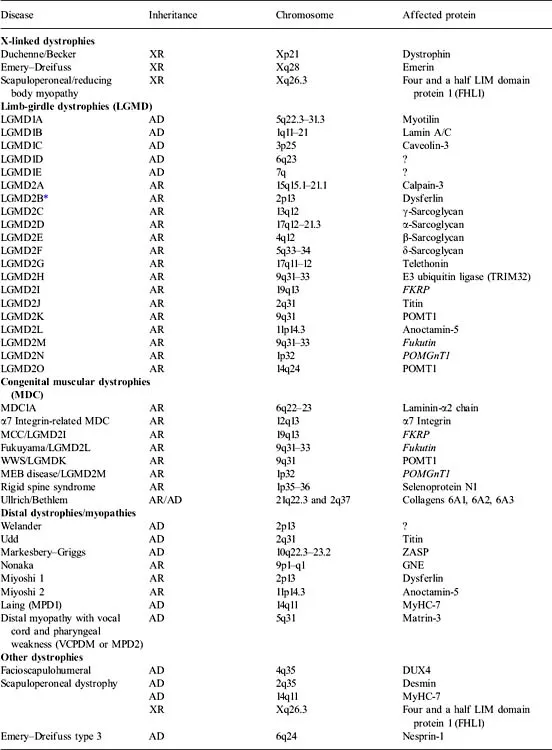
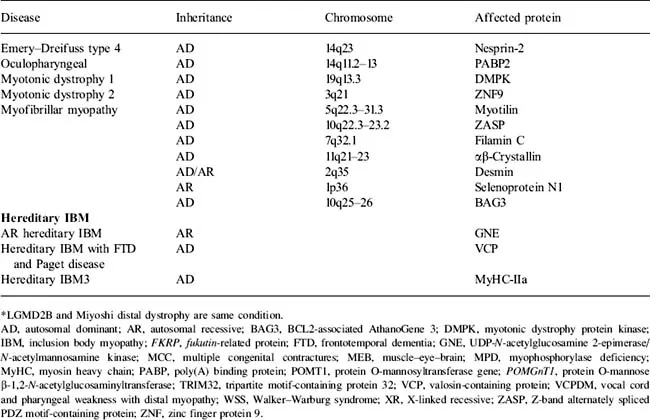
There are problems with this traditional classification of muscular dystrophies. We now know that most of the genetic defects previously found to be associated with congenital muscular dystrophy can all be associated with milder, adult-onset dystrophy (see Table 1.1). In addition, clinical heterogeneity is sometimes evident within family members with specific mutations such that some may manifest with a limb-girdle pattern of weakness, whereas other members in the family have distal weakness (e.g., Miyoshi myopathy, anterior tibial myopathy, LGMD2B are all associated with dysferlin mutations). Thus, it may be more appropriate to classify the dystrophies by the genetic defect (e.g., dysferlinopathies, calpainopathy) and to understand the specific clinical phenotype, including age of onset and patterns of weakness that may be associated with the specific disorders. However, the classic terminology (e.g., LGMD) is so ingrained that it is likely to persist until a new generation of clinical investigators armed with the explanations for divergent phenotypes writes its textbooks.
Molecular pathogenesis for dystrophies
The muscular dystrophies can be caused by mutations that encode for sarcolemmal, basement membrane, sarcomeric, nuclear structural proteins, or enzymes (Figures 1.1 and 1.2, Table 1.2). Further, some disorders are caused by mutations that affect splicing of mRNA (e.g., the myotonic dystrophies) or by yet still unknown mechanisms (e.g., FSHD). The pathogenesis of the various forms of muscular dystrophy will be discussed in subsequent chapters.
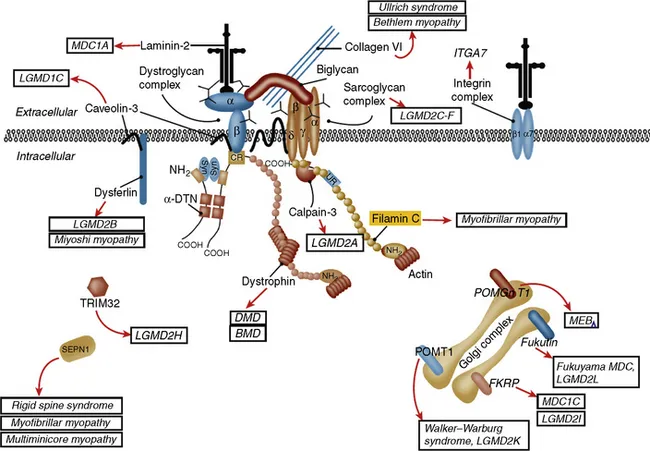
Figure 1.1 Sarcolemmal membrane and enzymatic proteins. This schematic shows the location of various sarcolemmal and enzymatic proteins associated with muscular dystrophies. The diseases caused by these molecules when mutated are shown in boxes. Dystrophin, via its interaction with the dystroglycan complex, connects the actin cytoskeleton to the extracellular matrix. Intracellularly, it interacts with dystrobrevin (α-DTN) and syntrophins (Syn) (shown in blue). Extracellularly, the sarcoglycan complex (orange) interacts with biglycan, which connects this complex to the dystroglycan complex and the extracellular matrix collagen. Intracellularly, δ- and γ-sarcoglycans interact with filamin C. The four proteins shown in the Golgi complex have been demonstrated to affect the glycosylation state of the α-dystroglycan and mediate its binding to the extracellular matrix. Fukutin and fukutin-related protein (FKRP) have been shown to localize to the medial Golgi. The localization of POMT1 (protein O-mannosyltransferase), POMGnT1 (protein O-linked mannose β-1,2-N-acetylglucosaminyltransferase), and LARGE (another putative glycosyltransferase) is unknown but believed to be in the Golgi complex as these enzymes are involved in the glycosylation process. BMD, Becker muscular dystrophy; DMD, Duchenne muscular dystrophy; ITGA7, integrin-α7; LGMD, limb-girdle muscular dystrophy; MDC, congenital muscular dystrophy; MEB, muscle–eye–brain; SEPN, selenoprotein N; TRIM, E3 ubiquitin ligase.
(From Amato AA, Russell J (2008) p. 530. Neuromuscular Disease. McGraw-Hill, New York, with permission.)
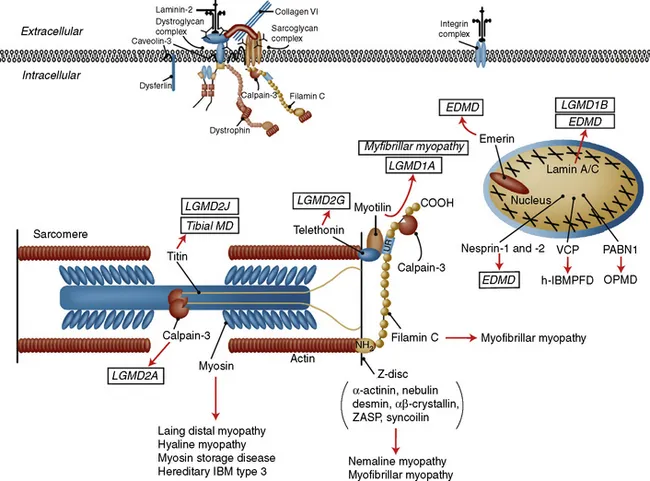
Figure 1.2 Sarcomeric and nuclear proteins involved in the muscular dystrophies. The schematic for the sarcomere and the nucleus showing the localization of the proteins involved in muscular dystrophies. The diseases they give rise to are shown in boxes. EDMD, Emery–Dreifuss muscular dystrophy; h-IBMPFD, hereditary inclusion body myopathy with Paget disease of bone; IBM, inclusion body myopathy; LGMD, limb-girdle muscular dystrophy; MD, muscular dystrophy; OPMD, oculopharyngeal muscular dystrophy; PABN, polyadenylate binding nuclear protein; VCP, valosin-containing protein; ZASP, Z-band alternately spliced PDZ motif-containing protein.
(From Amato AA, Russell J (2008) p. 531. Neuromuscular Disease. McGraw-Hill, New York, with permission.)
Table 1.2 Molecular defects associated...
Table of contents
- Cover
- Contents
- Series Editors
- Copyright
- Handbook of Clinical Neurology 3rd Series
- Foreword
- Preface
- List of Contributors
- Chapter 1: Overview of the muscular dystrophies
- Chapter 2: Dystrophinopathies
- Chapter 3: Sarcoglycanopathies
- Chapter 4: Congenital muscular dystrophies
- Chapter 5: The collagen VI-related myopathies
- Chapter 6: Limb-girdle muscular dystrophy 2A
- Chapter 7: Dysferlinopathies
- Chapter 8: Other limb-girdle muscular dystrophies
- Chapter 9: Limb-girdle muscular dystrophy 2H and the role of TRIM32
- Chapter 10: Caveolinopathies
- Chapter 11: Myofibrillar myopathies
- Chapter 12: Emery–Dreifuss muscular dystrophy
- Chapter 13: Facioscapulohumeral dystrophy and scapuloperoneal syndromes
- Chapter 14: Oculopharyngeal muscular dystrophy
- Chapter 15: Myotonic dystrophy types 1 and 2
- Chapter 16: Distal muscular dystrophies
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Muscular Dystrophies by Robert C. Griggs,Anthony A. Amato in PDF and/or ePUB format, as well as other popular books in Medicine & Neurology. We have over 1.5 million books available in our catalogue for you to explore.