Growth hormone (GH) has been used therapeutically for over 50 years. Since the development of a nearly unlimited supply of recombinant human GH in the mid-1980s, children with less severe GH deficiency can also profit from GH replacement therapy. Careful and accurate diagnosis and specific dosing, both essential to ensure normal height development, require the clinician to understand the finer points of clinical trials, to acquire quality evidence and assess the benefits of therapeutic intervention. Furthermore, genetic and environmental factors influencing GH sensitivity and responsiveness need to be taken into account. In this second edition all these aspects are covered in depth. Clinical examination, detailed auxological measurements, bone age assessment, molecular analysis and neuro-radiological evaluation as well as an adaptive strategy of dosing focusing on a patient's individual responsiveness are discussed in detail. This volume of Endocrine Development is essential reading for pediatric endocrinologists, pediatricians and clinical nurse specialists involved in GH therapy.

- 138 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Current Indications for Growth Hormone Therapy
About this book
Information
Topic
MedicineChapter 7
Hindmarsh PC (ed): Current Indications for Growth Hormone Therapy, ed 2, revised.
Endocr Dev. Basel, Karger, 2010, vol 18, pp 92-108
Endocr Dev. Basel, Karger, 2010, vol 18, pp 92-108
______________________
Current Indications for Growth Hormone Therapy for Children and Adolescents
Erick Richmonda · Alan D. Rogolb
aPediatric Endocrinology, National Children's Hospital, San José, Costa Rica; bPediatric Endocrinology, Riley Hospital, Indiana University School of Medicine, Indianapolis, Ind., and University of Virginia, Charlottesville, Va., USA
______________________
Abstract
Growth hormone (GH) therapy has been appropriate for severely GH-deficient children and adolescents since the 1960s. Use for other conditions for which short stature was a component could not be seriously considered because of the small supply of human pituitary-derived hormone. That state changed remarkably in the mid-1980s because of Creutzfeldt-Jakob disease associated with human pituitary tissue-derived hGH and the development of a (nearly) unlimited supply of recombinant, 22 kDa (r)hGH. The latter permitted all GH-deficient children to have access to treatment and one could design trials using rhGH to increase adult height in infants, children and adolescents with causes of short stature other than GH deficiency, as well as trials in adult GH-deficient men and women. Approved indications (US Food and Drug Administration) include: GH deficiency, chronic kidney disease, Turner syndrome, small-for-gestational age with failure to catch up to the normal height percentiles, Prader-Willi syndrome, idiopathic short stature, SHOX gene haploinsufficiency and Noonan syndrome (current to October 2008). The most common efficacy outcome in children is an increase in height velocity, although rhGH may prevent hypoglycemia in some infants with congenital hypopituitarism and increase the lean/fat ratio in most children - especially those with severe GH deficiency or Prader-Willi syndrome. Doses for adults, which affect body composition and health-related quality of life, are much lower than those for children, per kilogram of lean body mass. The safety profile is quite favorable with a small, but significant, incidence of raised intracranial pressure, scoliosis, muscle and joint discomfort, including slipped capital femoral epiphysis. The approval of rhGH therapy for short, non-GH-deficient children has validated the notion of GH sensitivity, which gives the opportunity to some children with significant short stature, but with normal stimulated GH test results, to benefit from rhGH therapy and perhaps attain an adult height within the normal range and appropriate for their mid-parental target height (genetic potential).
Copyright © 2010 S. Karger AG, Basel
Introduction
Human growth hormone (hGH) is a single-chain, 191-amino-acid protein of 22 kDa molecular weight. It is synthesized, stored and released from the somatotropes of the anterior pituitary gland. Multiple additional molecular variants, including a 20-kDa form produced by the gene deletion of 14 amino acids and other posttranslational isoforms, for example glycosylated and sulfated forms, of unknown physiological significance exist within the systemic circulation [1].
GH is relatively species-specific since only primate GH has efficacy in the human [2, 3]. None of the small trials with animal GH or enzymatically-produced fragments from animal GH showed growth-promoting efficacy in the human [4].
hGH was first extracted in the late 1940s [5] and administered to humans beginning in 1958 [6, 7]. Treatment of both children and adults was reported in that first clinical trial, although the latter took a number of years for confirmation of efficacy since so little hormone was available (essentially one pituitary per child per day). Thus, in the early years only profoundly GH-deficient children received treatment. Much of the time so little hormone was available that many children were treated 6 or 9 months a year permitting other, equally needy, children to reap some benefit from hGH therapy.
That all changed in the early 1980s when it became clear that Creutzfeldt-Jakob disease could be transmitted by human brain tissue and recombinant (r)hGH became available [8]. At that time a seemingly endless supply of the pure 22-kDa hormone permitted all GH-deficient children to have access to treatment and one could now design trials using rhGH to increase adult height in infants, children and adolescents with causes of short stature other than GH deficiency, as well as trials in adult GH-deficient men and women. The next 20 years led to at least 7 additional indications in children and 3 new ones in adults (table 1).
hGH is administered to promote linear growth in short children. The following are the US Food and Drug Administration (FDA)-approved indications for GH (current to October 2008) and in parentheses the indications approved in Europe (E) by the European Agency for the Evaluation of Medicinal Products (EMEA).
The most common efficacy outcome in infants, children and adolescents is an increase in linear growth, although rhGH may prevent hypoglycemia in some infants with congenital hypopituitarism and increase the lean/fat ratio in children with the Prader-Willi syndrome (see below).
The dose-response curve for height gain versus dose of rhGH (log scale) is rather flat in those with GH deficiency [9], even through the much higher doses (including 100 μg/kg/day) administered more recently [10]. The dose for adults which affects body composition and health-related quality of life are much lower per kilogram of body mass.
Table 1. Approved indications for GH therapy in children and adults in the USA and Europe (E)
| Children |
| Growth hormone deficiency (E) |
| Chronic kidney disease (E) |
| Turner syndrome (E) |
| Small-for-gestational age infants who fail to catch up to the normal growth percentiles (E) |
| Prader-Willi syndrome (E) |
| Idiopathic short stature |
| SHOX gene haploinsufficiency (E) |
| Noonan syndrome |
| Adults |
| Growth hormone deficiency (E) |
| HIV/AIDS wasting |
| Short bowel syndrome |
In this review, the studies that were selected for analysis were mainly randomized controlled studies, with the larger number of patients and the longest treatment duration of the available publications.
Growth Hormone Deficiency (GHD)
The administration of GH to treat children with sort stature resulting from GHD or GH insufficiency has now accrued over 40 years of clinical experience. In 1985, the US FDA approved the use of rhGH for the treatment of GHD and remains the primary indication for GH treatment in childhood. GHD is fundamentally a clinical diagnosis, based upon auxologic features. Assessment of laboratory tests, whether static, for example the measurement of IGF-1 and IGFBP-3, or dynamic, for example, secretagogue-stimulated GH secretion is confirmatory [11]. Radiologic evaluation of the hypothalamus and pituitary (CT scan or MRI) is helpful in patients with suspected congenital GHD or to detect space-occupying lesions (see Chapters 1 and 3 for fuller discussion of these points).
The primary objectives of therapy of GHD are normalization of height during childhood and attainment of adult height within the normal range. The greater efficacy demonstrated in recent large international databases [12, 13] compared with the initial studies of GH treatment in GHD patients (table 2) probably reflects the combined effects of the higher GH dose, the more physiologic injection frequency, and the younger age at initiation of treatment. Current consensus guidelines recommend a dose in the range of 0.025-0.05 mg/kg/day [21, 22]. Under special circumstances, higher doses may be required, including adolescents with late diagnosis and diminished period of time for catch-up growth [23]. Recently it has been proposed that IGF-1-based GH dosing may improve growth responses, although at higher average GH doses [24].
Table 2. GH trials in children with GHD
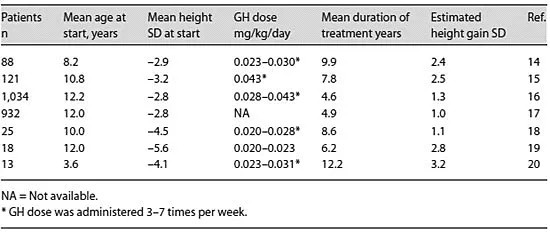
Significant side effects of GH treatment in children are uncommon. These include idiopathic intracranial hypertension, prepubertal gynecomastia, arthralgia, and edema [21, 22]. Management of these side effects may include either transient reduction of dosage or temporary discontinuation of GH. In the absence of other risk factors, there is no evidence that the risk of leukemia, brain tumor recurrence, slipped capital femoral epiphysis or diabetes are increased in recipients of long-term GH treatment.
GHD may or not persist into adulthood. After the...
Table of contents
- Cover Page
- Front Matter
- Clinical Trials: Planning and Analysis
- The Evidence Base for Growth Hormone Effectiveness in Children
- Safety of Recombinant Human Growth Hormone
- Diagnosis of Growth Hormone Deficiency
- Biological Determinants of Responsiveness to Growth Hormone: Pharmacogenomics and Personalized Medicine
- Clinical Considerations in Using Growth Hormone Therapy in Growth Hormone Deficiency
- Current Indications for Growth Hormone Therapy for Children and Adolescents
- GH Use in the Transition of Adolescence to Adulthood
- Author Index
- Subject Index
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Current Indications for Growth Hormone Therapy by P. C. Hindmarsh in PDF and/or ePUB format, as well as other popular books in Medicine & Alternative & Complementary Medicine. We have over 1.5 million books available in our catalogue for you to explore.