![]()
CHAPTER 1
Different Flavours of Fragments
CHRIS ABELL*a AND CLAUDIO DAGOSTINa
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
*E-mail:
[email protected] 1.1 Fragment-Based Drug Discovery
Fragment-based drug discovery is becoming a powerful technology in the arsenal of the pharmaceutical companies to aid discovery of new small-molecule therapeutics, establish the druggability of biological targets, and discover alternative inhibition sites on already established ones.1,2 With the market approval of Zelboraf® (Vemurafenib – the first drug discovered via fragment-based drug discovery3) the approach has a measure of validation and interest in the field continues to grow.
Fragments are small molecules that may become parts of a larger molecule, but in some cases were fragments of known drugs, that have been used as starting points to find new inhibitors for different biological targets. A fragment is a small, typically aromatic, organic molecule of molecular weight <250 Da, which is very soluble and chemically stable. The use of such small molecules at the beginning of a medicinal chemistry program allows an effective exploration of chemical space and a more rational design of the final molecule through iterations guided by structural knowledge of the site of binding. In the best cases, the approach can offer a less expensive and faster route to potent and better quality lead compounds compared with more traditional approaches such as high-throughput screening (HTS) of higher molecular weight compounds.
1.2 Different Types of Fragments
Views on the composition of fragment libraries have evolved over time. Some libraries were assembled by taking known drugs and dissecting them into fragments4 or by computationally finding diverse fragments.5 It was apparent from the start that solubility would be very important because of the concentrations used for screening (often tens of mM). Another key criterion was lack of reactive groups6 although current interest in covalent inhibitors is beginning to impact on thinking here.7 Fragment libraries, although generally diverse, were mainly composed of flat aromatic or heteroaromatics. Astex introduced the Rule of 38 to map across to the Lipinski Rule of 5, and soon after, commercial Rule of 3 compliant libraries became available.
There remain different views on the best size for a fragment library. Some organisations have libraries of over 20 000 fragments whereas others use small libraries (for a recent study of an NMR library see ref. 9). We typically screen 800–1300 compounds (generally by thermal shift assay), from which we identify 20–100 fragments for further screening by ligand-based NMR. Structure–activity relationship (SAR) analysis is then conducted around the NMR hits by buying or making similar compounds. At Astex, similar sized libraries are screened by X-ray crystallography and NMR spectroscopy.10 In large pharmaceutical companies there has been a tendency to screen larger fragment libraries, e.g., by surface plasmon resonance (SPR); an advantage being that SARs will emerge from the initial screen. A consideration in deciding how many fragments to screen is the expected hit rate. This can be very high (20%+) in the case of a kinase screen or vanishing low when looking for compounds binding to a surface site to disrupt a protein–protein interaction.11
Fragment libraries have been designed based on natural products e.g., “fragments of life” libraries including highly soluble metabolites, natural product derivatives and biaryl molecules designed to mimic the architectural motifs of proteins (α, β and γ turns).12 This library was tested against leukotriene A4 hydrolase by screening about 200 fragments using X-ray crystallography. Thirteen hits were obtained of which 11 bound to the active site, while 2 biaryls were found binding in a shallow pocket on the surface of the protein.
In another report, the Waldmann group reported a chemoinformatic analysis of more than 180 000 natural product structures that was used to obtain about 750 000 fragments of different Mw and properties rich in sp3 centres.13 This virtual library was then filtered according to an HTS filter and Rule of 3 derived parameters and re-ordered in 2000 clusters. Part of the library (193 fragments) was tested on p38α MAP kinase using biochemical assays, leading to 12 hits being identified with potencies between 6 and 0.4 mM and ligand efficiencies (LEs) ranging from 0.19 to 0.28. Most of the hits predictably bound to the hinge region of the kinase ATP site, but interestingly there were some that bound to an allosteric site, as a novel class of type-III inhibitors. The library was also tested on a less tractable class of targets, the tyrosine phosphatases, including Mycobacterium tuberculosis protein tyrosine phosphatase A and B. The best hits had an IC50 of <100 µM and LEs of almost 0.50. However, despite the growing interest in 3D fragments there is no compelling evidence yet that this will result in more or better hits.14
1.3 How We Identify Fragments
Given their size, fragments generally bind very weakly (100 µM to 10 mM) to biological targets. Consequently, there is a particular emphasis in the biophysical techniques to detect binding.15 These include NMR (ligand- and protein-based), X-ray crystallography, SPR, isothermal titration calorimetry (ITC), thermal shift, and native mass spectrometry. These different approaches mean that the start of a fragment-based drug discovery (FBDD) project can be quite different in different labs, but the end game is generally similar. The screening approach used is partly dictated by available facilities, and partly by organisational constraints, although in all the cases, a single technique is rarely used in isolation. Typically a combination of techniques is employed in order to minimise false positives and false negatives and thus provide reliable chemical starting points for subsequent design and synthesis.
Virtual screening using in silico docking methods has been used, but usually in support of subsequent biochemical and biophysical assays. The different binding modes of simple fragments may only exhibit small differences in free energy of binding compared to larger compounds, so the inaccuracy inherent in scoring functions does not allow a reliable ranking of fragment-binding modes.16
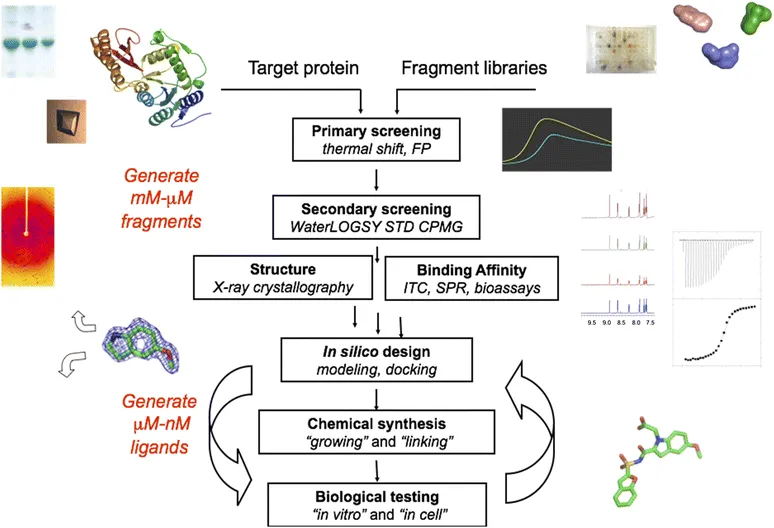
In our lab, the first screen generally employs the Sypro Orange fluorescence-based thermal shift screen (Figure 1.1);17 where the selected hits are those giving the largest increase in thermal stability of the protein. This offers a quick way to identify 50–100 compounds to proceed on to NMR screening. However, the shifts are generally small and the screen should be repeated to improve the reliability of the hits. This is a very simple application of this screen, and it is clear that in more complex systems, e.g., when studying protein–protein interactions, good hits can give even significant negative shifts. A recent study on Checkpoint kinase 2 used the AlphaScreen kinase biochemical assay and a thermal shift screen.18 A good correlation was found for compounds which were hits in both these orthogonal screens. Such high-concentration biochemical screens can give a high frequency of false positives so it is important to do a counter-screen to check for interfering compounds.
Figure 1.1 Fragment screening cascade.
In many companies, SPR has become the primary screening technique to identifying fragment hits. It has advantages of ease of setting up the assay, relatively high throughput, and requires low amounts of protein compared to other techniques (e.g., NMR and X-ray) used. The methodology is likely to become more popular as the cost of the instrumentation decreases.19 A later chapter by Giannetti is dedicated to this approach.
NMR techniques1,20,21 are among the most powerful and widely used to detect binding of fragments to target proteins. Ligand-based and protein-based methods each have advantages and limitations, and can complement each other. However it is the ligand-based methods that have found most general application. In our lab we use a combination of three such techniques (described in detail in Krimm’s chapter): WaterLOGSY, STD and CPMG. WaterLOGSY detects binding through transfer of magnetisation from the water molecules to the ligand bound to the protein. STD uses a pulse to saturate signals from methyl groups in the protein. The disturbance in magnetisation is then transferred through an NOE to the bound ligand, with the resulting difference spectrum only showing a peak of the ligand when this binds to the protein target. Finally CPMG, is a relaxation-time-edited NMR experiment that exploits differences in transverse relaxation time (T2). Proteins (and bound ligands) have a small T2 while free ligands have a large T2. Thus monitoring T2, binding can be detected when the signal of the ligand decreases.
In a given screen, some fragments show binding by all three techniques (e.g., Figure 1.2), some by only two, some by one. We progress preferentially those giving a positive outcome from all three experiments. An example of the power of fragment screening by NMR was in a study on Pin1. After a million-compound HTS failed to give any hits, an NMR screen of 1200 fragments using the three 1D ligand-based techniques identified 5 hits from 4 different series and yielded a lead compound of 260 nM IC50.22 Astex used a multistage NMR screen against HSP90 to select 125 compounds from a 1600 fragment library for X-ray crystallography. Fragments that bound to the protein were initially identified in a WaterLOGSY experiment. The presence of 200 µM ADP made it possible to identify those compounds that bound to the nucleotide site from their effect on the ADP signal. When Mg2+ was then added, this increased the affinity of HSP90 for ADP 20-fold, and led to displacement of those frag...