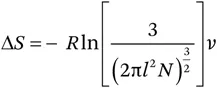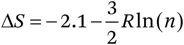Disulfide bonds are unique among post-translational modifications, as they add covalent crosslinks to the polypeptide chain. Accordingly, they can exert pronounced effects on protein folding and stability. This is of particular importance for secreted or cell surface proteins, where disulfide bonds are abundant and serve to stabilize proteins against unfolding and dissociation in the extracellular milieu. However, in addition to these bonds providing security to a natively folded protein or aiding the folding process by stabilizing folding intermediates, the cysteines that form these bonds can be perilous during the maturation of nascent polypeptide chains as they enter the endoplasmic reticulum where the concentration of unfolded proteins approaches millimolar levels. This danger is even greater if the native bonds ultimately form between non-consecutive cysteines that are distant in the linear sequence or if non-native bonds are a prerequisite to achieving the final, functional structure of a protein. A wealth of exquisite detail has been obtained from in vitro studies on the biophysical effects of disulfide bonds on protein folding. Correspondingly, in-depth in vivo studies have established that the same principles apply to oxidative folding in a cell, but reveal a much more complex folding trajectory for many of the proteins that have been examined. In this chapter, we review the biophysical properties of disulfide bonds and how they affect the structure and folding of individual proteins. Based on this, we discuss similarities and differences between in vitro and in vivo folding reactions. The types of disulfide bonds that form during co-translational protein folding are described, as are the cellular strategies for accommodating this risk-laden covalent modification. We conclude with a discussion of the impact of disulfide bonds on protein misfolding and human disease.
1.1.1Stabilization of Proteins by Disulfide Bonds
Early protein folding studies were interpreted as suggesting that the native state of a protein corresponds to one well-defined conformation, whereas the unfolded state corresponds to a random coil.1 If no other states than either the native or unfolded state are kinetically or thermodynamically stable, we speak of a two-state folding mechanism for a protein.2 This scenario set the stage for early analyses of the role that disulfide bonds play in protein stability.
An ideal random coil is devoid of any long-range interactions except excluded volume effects. It behaves as a freely joined chain with segments of defined length.3 In such a system, the impact of a covalent crosslink between two defined residues of the polypeptide chain, such as a disulfide bond, would be greatest on the unfolded state, significantly decreasing the conformational freedom of the random coil. Restricting the conformational space of the unfolded state reduces its entropy. Hence, in the presence of a disulfide bond, the entropy change for the reaction to the ordered native state is less negative, with net stabilization of the folded protein as a result. This model will be called the chain-entropy model in the following sections. A quantitative description was developed by Flory,4 Schellman5 and Poland and Scheraga.6 The decrease in entropy of the unfolded state is derived from the probability that two otherwise free elements of the chain are now found in a defined volume element (v). The mathematical description of the problem, based on polymer theory, can be found in the equation
where R is the gas constant and l the average length of a statistical segment of the chain composed of N segments; in proteins, l is assumed to be 3.8 Å, corresponding to one amino acid. A major point of discussion has been the suitable choice of v. A value of 57.9 Å3 based on the closest possible approach of two thiols is mostly in use.7 Hence eqn (1.1.1) can be simplified to
where n is the number of amino acids bridged by the disulfide bond. Based on a study of ribonuclease (RNase) T1 with no, one and two intact disulfide bonds, eqn (1.1.2) was developed by Pace et al.7 They not only found a good correlation between n and ΔΔG upon removal of disulfide bonds in RNase T1, but also observed agreement between the predictions from these equations and the experimental data for lysozyme, RNase A and the antibody CL domain.
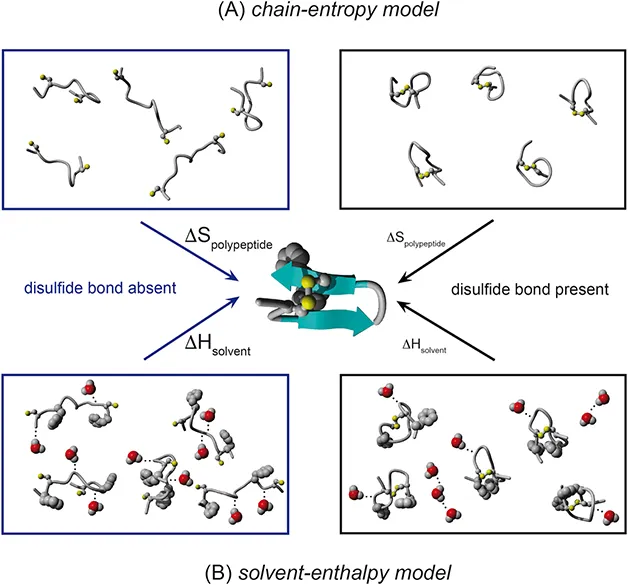
The above equations have two main consequences. Conceptually, the stabilization of a protein is thought to be an entirely entropy-driven process with an impact exclusively on the unfolded state. In theory, the stabilization achieved by a disulfide bond should therefore always increase with increase in the number of amino acids between the two cysteines. Despite its appealing simplicity, in practice this theory falls short in important aspects of real proteins. It treats the unfolded polypeptide chain as a system devoid of any intra- or intermolecular interactions. Furthermore, the water surrounding a protein is an important factor in shaping the free energy landscape of the polypeptide chain, and also in the native state, but is neglected in the equations.8–10 The impact of these considerations regarding disulfide bonds was addressed by Doig and Williams in 1991.11 They argued that disulfide bonds may significantly decrease the solvent-accessible surface in the unfolded state of a protein. As a consequence, hydrophobic residues and also hydrogen-bond donors and acceptors may become buried. Burial of hydrophobic residues would lead to less ordering of water and thus a higher entropy of the solvent surrounding disulfide-containing proteins. Consequently, the hydrophobic effect, a major driving force in protein folding, should be less pronounced. On the other hand, hydrogen bonding with the solvent will be less extensive for a more compact unfolded state, thus reducing this competition. As a result, folding to the native state will be enthalpically more favorable. According to the authors, this enthalpic contribution must be considered as the major stabilizing factor of disulfide bonds. This model will therefore be called the solvent-enthalpy model in the following sections. As in the chain-entropy model, effects on the native state are also neglected in this model. Both models are summarized in Figure 1.1.1.
Figure 1.1.1 Models for the role of disulfide bonds on polypeptide stability. (A) The chain-entropy model predicts a smaller change in entropy (ΔS) upon folding of a polypeptide chain containing a disulfide bond than of one lacking a disulfide bond. This leads to a net stabilization of the native state. (B) The solvent-enthalpy model predicts fewer solvent–polypeptide interactions (water molecules are displayed in a CPK representation and hydrogen bonds as dashed lines) and less exposure of hydrophobic residues for a polypeptide chain containing a disulfide bond than for a polypeptide chain lacking one. This is assumed to reduce the enthalpy change (ΔH) upon loss of solvent–polypeptide interactions during folding and thus will lead to net stabilization of the native state.
A variety of experimental evidence argues either for or against these two theories, rendering the problem much more complex, but the data obtained from the various experiments also provide a chance for further insights. The advent of site-specific mutagenesis offe...