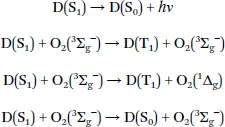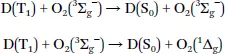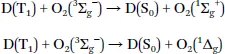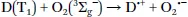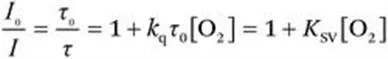Sergey M. Borisova
1.2 Mechanism of Oxygen Quenching
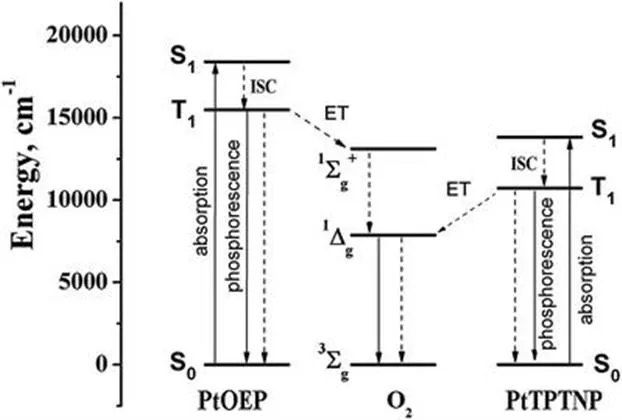
Oxygen is one of the most powerful luminescence quenchers. Quenching of fluorescent dyes (excited singlet state, S1) and phosphorescent dyes (excited triplet state, T1) is spin-allowed. Moreover, the energies of excited states of oxygen (1∑g+ and 1Δg) are lower than the energies of the excited states of most organic dyes and metal complexes (Figure 1.1), which makes quenching via energy transfer favourable.
Figure 1.1 Energy diagram for two phosphorescent oxygen indicators: platinum(ii) octaethylporphyrin (PtOEP) and platinum(ii) tetraphenyltetranaphthoporphyrin (PtTPTNP).
The mechanism of oxygen quenching is rather complex and the exact pathways and formed products depend on many factors.1 Electron-exchange Dexter-type energy transfer is the predominant mechanism of oxygen quenching. Quenching of fluorescent dyes (D) can result in the formation of the dye in the triplet excited state or in the ground state:1
The triplet state of the dye is deactivated to the ground state:
For the quenching of phosphorescence, the dye is deactivated into the ground state and singlet oxygen is formed:
Depending on the triplet energy of the dye, formation of singlet oxygen either only in the 1Δg state (e.g. for PtTPTNP, Figure 1.1) or in both 1Δg and 1∑g+ states (e.g. for PtOEP) is possible. Notably, O2(1∑g+) deactivates very fast into O2(1Δg) state.
Apart from the energy transfer, electron transfer leading to superoxide is also possible:
This process can play a significant role for metal complexes with strong reducing properties (particularly in the excited state), for instance Ir(iii) cyclometalated complexes.2 Rapid back electron transfer can result in the formation of singlet oxygen and the sensitizer in the ground state.
Importantly for all these processes, singlet oxygen represents one of the main products. Since its deactivation to the triplet state regenerates the analyte, optical oxygen sensors do not consume the analyte in theory. However, the lifetime of singlet oxygen in polymers can be much longer compared to that in the aqueous phase (∼3 µs), which can be sufficient for it to react with the sensor components (dye or polymer), see Chapter 1.7.
Independent of the quenching mechanism, the quenching behavior for dissolved dyes is described by the Stern–Volmer equation:
where I0(τ0) and I(τ) are the luminescence intensity (decay time) in the absence and in the presence of oxygen, respectively, kq is the bimolecular quenching constant and KSV is the Stern–Volmer constant.
From eqn (1.1) it is evident that the efficiency of quenching depends on both the bimolecular quenching constant and the decay time of the luminophore τ0. The kq constant is determined mostly by oxygen diffusion since the diffusion of the much larger dye is significantly slower already in solution and is virtually non-existant for immobilized dyes. The kq constant often approaches the diffusion-controlled limit kdiff for quenching of fluorescence1 but is lower for quenching of phosphorescence. For common phosphorescent indicators such as Pt(ii) porphyrins or Ru(ii) polypyridyl complexes, it is usually close to 1/9 of kdiff,2 where 1/9 is the spin statistical factor accounting for the formation of both products in the singlet state. However, kq is sometimes higher than this value even in the case of purely energy transfer-based quenching1 and may be even higher if electron transfer is involved.2
Clearly, the τ0 has a much stronger influence on the Stern–Volmer constant than kq. In fact, assuming a kq = kdiff = 2.1 × 1010 M−1 s−1 for an air-saturated toluene solution (C(O2) ≈ 1.8 mM) of a typical fluorescent dye with τ0 of 4 ns, the I0/I value calculated with eqn (1.1) is only 1.15. On the other hand, for a phosphorescent indicator such as PtOEP (τ0 = 85 µs) even with much lower kq 2.4 × 109 M−1 s−1 (1/9kdiff) the luminescence intensity and decay time decrease 367-fold in the same conditions. Since the diffusion of oxygen is significantly slower in the polymers compared to the solution, it is evident that only phosphorescent indicators will provide the required resolution when embedded in common polymeric matrices. Additionally, whereas the tunability of fluorescence decay times is usually limited by 1–2 orders of magnitude, the phosphorescence decay time can vary from several microseconds to hundreds of milliseconds. This provides virtually unlimited flexibility in designing oxygen-sensing materials for very different applications.