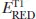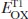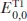![]()
Chapter 1
Ketones and Aldehydes
Shin Kamijo
Graduate School of Sciences and Technology for Innovation, Yamaguchi University, Yamaguchi, Japan
1.1Introduction
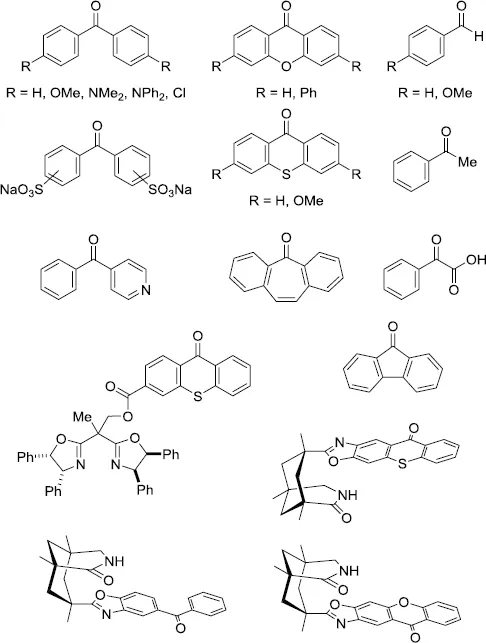
Extensive studies have been carried out in the photochemistry of carbonyl compounds, especially ketones, since photoexcited ketones are known to induce various types of transformations [1–4]. Among them, one of the most significant features of aryl ketones, such as benzophenone (Ph2CO) and its derivatives, is their ability to act as photoorganocatalysts (POCs) (Scheme 1.1).
For instance, Ph
2CO is readily excited to a singlet state with light irradiation [>350 nm for
n–
π*] and is then rapidly converted to a triplet state through intersystem crossing. The relatively longer lifetimes of the triplet state of aryl ketones [10 ns–1
μs for Ph
2CO depending on the solvent used] have allowed the promotion of photochemical transformations
via energy transfer (EnT), hydrogen-atom transfer, and electron transfer (ET) reactions [redox potentials for triplet Ph
2CO:
+ 1.28 V vs SCE,
− 0.61 V vs
SCE; and energy for triplet Ph
2CO:
3.0 eV]. This chapter mainly covers recent representative catalytic transformations involving carbonyl compounds, including ketones and aldehydes, under photoirradiation conditions.
Scheme 1.1.Representative ketone and aldehyde POCs.
1.2Functionalizations of Unsaturated Bonds
1.2.1Alkylation of olefins
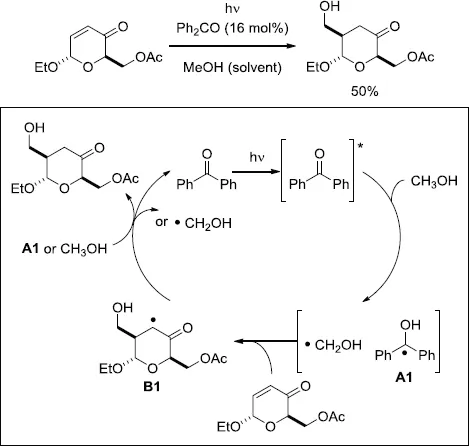
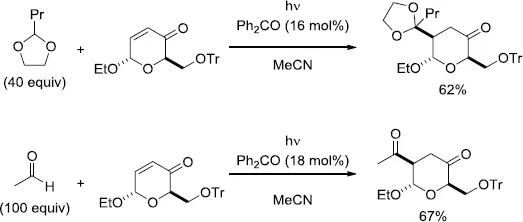
One of the pioneering investigations on the alkylation of oxygen containing compounds, such as alcohols and acetals, via the photoinduced conjugate addition to carbohydrate-derived enones in the presence of a catalytic amount of benzophenone (Ph2CO) was carried out by Fraser-Reid and co-workers [5–7]. As an example, irradiation of the enone derived from acetylated glucose in MeOH containing a catalytic amount of Ph2CO (16 mol%) resulted in the regioselective formation of the 1,4-adduct in 50% yield (Scheme 1.2) [5]. The reaction was proposed to proceed with hydrogen-atom abstraction from MeOH by the photoexcited Ph2CO. The derived hydroxymethyl radical then added to the enone in 1,4-fashion from the less-hindered face. This addition step should be favored due to the nucleophilic nature of the hydroxymethyl radical. Subsequently, the hydrogen-atom transfer between the generated radical B1 and the ketyl radical A1 or MeOH took place to form the adduct [6]. In addition to MeOH, the applicable substances were expanded to include diols, acetals, and aldehydes (Scheme 1.3) [7]. A similar type of diastereoselective alkylation of isopropanol, 1,3-dioxolane, and tetrahydrofuran (THF) was also investigated by Mattay and co-workers [8].
Scheme 1.2.Addition of alcohols to cyclic enones.
Scheme 1.3.Addition of acetals or aldehydes to cyclic enones.
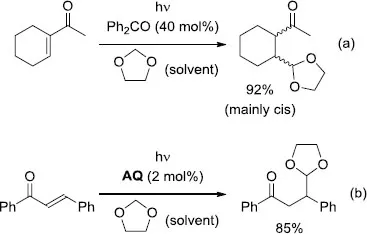
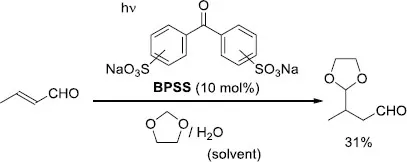
The research group of Fagnoni and Albini achieved the catalytic photoinduced alkylation of 1,3-dioxolane with α,β-unsaturated ketones (aliphatic, both acyclic and cyclic, as well as aryl-substituted) in the presence of a ketone as POC [9]. The irradiation of the alkyl ketone in dioxolane with Ph2CO (40 mol%) led to the formation of the addition product with a yield of 92% (Scheme 1.4(a)). On the other hand, the reaction employing the aryl ketone substrate was efficiently catalyzed by anthraquinone (AQ, 2 mol%) rather than Ph2CO, furnishing the expected adduct in 85% yield (Scheme 1.4(b)). Further investigations enabled the alkylation of dioxolane using α,β-unsaturated aldehydes in aqueous solution [10], as well as by designing the water-soluble photocatalyst disodium benzophenonedisulfonate (BPSS) (Scheme 1.5) [11].
Scheme 1.4.Addition of 1,3-dioxolane to enones.
Scheme 1.5.Addition of 1,3-dioxolane to enals.
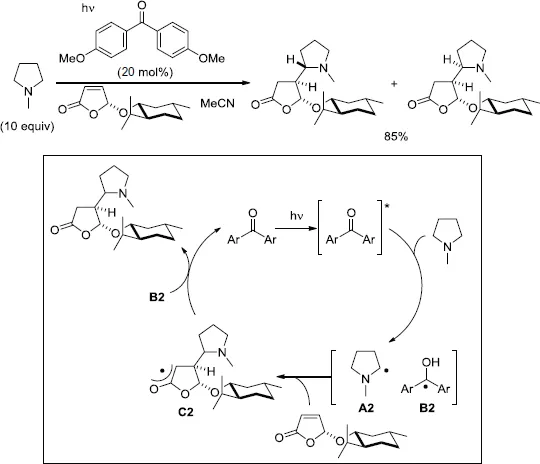
Hoffmann and co-workers investigated intensively the diastereoselective alkylation of amine derivatives by a chiral furanone via a photoinduced conjugated addition in the presence of a catalytic amount of aryl ketones [12–16]. In the initial stage of the investigation, the alkylation of N-methylpyrrolidine was attained (Scheme 1.6) [12].
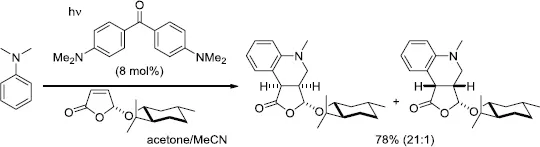
The treatment of the pyrrolidine (20 equiv) and the chiral furanone bearing a menthyloxy group in MeCN with a catalytic amount of 4,4′-dimethoxybenzophenone (10 mol%) under irradiation produced the addition products in 85% combined yield. A complete facial selectivity at the furanone ring was observed; however, the other asymmetric center adjacent to the nitrogen atom could not be controlled. The current strategy for diastereoselective alkylation of pyrrolidine derivatives was applied to the total synthesis of two pyrrolizidines alkaloids, viz. (–)-isoretronecanol and (+)-lauburine [13]. In the proposed catalytic cycle, the reaction was initiated by photoinduced electron transfer (PET) between the photoexcited aryl ketone and pyrrolidine followed by proton transfer to generate the α-aminoalkyl radical A2 and the ketyl radical B2 [14]. The derived radical A2 was added to the electron-deficient double bond of the furanone from the less-hindered face to form the oxoally radical C2. A hydrogen-atom transfer from the ketyl radical B2 to the radical C2 accounted for the recovery of the aryl ketone after the reaction. When dimethylaniline was employed as a starting amine in the presence of 4,4′-bis(N,N-dimethylamino)benzophenone (8 mol%), a radical tandem reaction took place to form tetrahydroquinolines in a highly diastereoselective manner (Scheme 1.7) [15]. Further investigations enabled the alkylation of aliphatic amines, including Et3N and six-membered azacycles with electron-deficient olefins [16].
Scheme 1.6.Diastereoselective addition of pyrrolidine to a furanone.
Scheme 1.7.D...