![]()
Chapter 1
Synthesis of Siloles (and Germoles) that Exhibit the AIE Effect
Joyce Y. Corey
Department of Chemistry and Biochemistry, University of Missouri–St. Louis, USA
1.1 Introduction
As reported in 2001 by Tang and co-workers [1], the identification of the aggregation-induced emission (AIE) effect was intimately connected with a study of siloles and stemmed from the simple observation that a wet spot containing 1-methyl-1,2,3,4,5-pentaphenylsilole (Ph4C4SiMePh) (MPS) on a thin-layer chromatographic (TLC) plate barely fluoresced under UV light but was clearly visible when the spot dried. In contrast to the expectation of the time period that in the solid state aggregation would cause quenching [aggregation-caused quenching (ACQ)] of the photoluminescence (PL) processes of luminophoric molecules, the silole exhibited the reverse behavior: a sample was virtually nonfluorescent in solution but fluorescent in the solid state. For applications in electronic devices, thin films of a luminophoric material are required and the appearance of ACQ would thus inhibit such applications. An electroluminescent (EL) device was constructed with MPS and was demonstrated to be a good light-emitting material for device applications [1]. The observation of AIE in siloles was not the beginning of the search for siloles that could be efficiently incorporated into organic light-emitting diodes (OLEDs), as Tamao et al. had demonstrated in 1996 that siloles were efficient electron-transporting materials and showed that the device performance was related to the substituents in the 2,5-positions of the ring [2]. This was actually the first demonstration that siloles could be good candidates as core components for organic EL devices. The AIE luminogens, however, have additional applications, for example, as chemical sensors, biological probes, and smart nanomaterials. The ‘turn-on/light-up’ nature of an AIE sensor makes for ready use in the field or for on-sight screening, to name just a couple of applications, which will be highlighted in other chapters.
This chapter focuses on the synthetic methods that have been used to target silacyclopentadiene and germacyclopentadiene derivatives (common names: silole and germole) and their related benzene-annulated derivatives. Synthetic examples that have actually been described as exhibiting the AIE effect will be discussed whenever possible. However, silole systems have been studied since the first report in 1959 by Braye and Hübel of the formation of Ph4C4SiPh2 (HPS), presumably by a rather obscure synthesis from reaction of Fe2(CO)6(PhC2Ph)2 with Ph2SiCl2, and related to that which was used to produce pentaphenylphosphole from the same iron precursor and PhPCl2 [3a]. The same group subsequently described a better method for the synthesis of HPS [3b] that is still in use today and is the first method that will be described in Section 1.3.1. The material covered will not provide a comprehensive listing of all systems that have been prepared but will attempt to describe the methodology that has been developed for targeting the silole core, illustrated with selected examples. Since three of the principal methods for silole formation predate the discovery of the AIE effect, testing for this phenomenon has understandably not been reported in publications prior to 2001. One of the more interesting examples was the first viable synthesis of HPS (in 1961), where it was stated: ‘In the solid state it exhibits a strong blue fluorescence in ultraviolet light…’ [3b]. It is this solid-state fluorescence that has become a feature of the AIE effect and proves once again that ‘chance favors the prepared mind’ in its discovery.
The rest of this chapter is divided into the following sections. Section 1.2 contains background material on the nomenclature of siloles and their benzene-annulated relatives and the numbering systems that are commonly used and that will be utilized in this chapter. A brief introduction to the bond formation methods in silicon chemistry that are relevant to the formation of siloles will be included, as this type of chemistry imposes certain limitations on how the synthesis of a particular silole might be approached. In general, it is the role of organic chemistry in forming the carbon portion of siloles and their related derivatives that has actually enabled the presence of various substitution patterns to be incorporated into the target metallacycle. This is seen particularly in the Tamao synthesis (Section 1.3.2) of siloles and in the formation of precursors to the benzene-annulated systems (Section 1.6.2). The substitution reactions possible at both the silicon center and the carbon centers of the metallacycle also play a role, particularly in the area of copolymer synthesis. Lastly, Section 1.2 gives brief comments on the early calculations for the HOMO and LUMO energy levels for siloles as compared with the related carbon parent and the heterocycles of N and S.
Section 1.3 contains the bulk of the synthetic chemistry and describes three basic approaches to siloles as well as the more recent methodology that involves the use of transition metals both stoichiometrically (early work) and catalytically. Section 1.4 covers methods that can be used to modify the silole core and 1.5 addresses whether the synthetic methods can be extended to the heavier Group 14 element, germanium. Section 1.6 will describe basic routes to silaindenes and silafluorenes that are related to those used for siloles. Section 1.7 contains a brief discussion on the extension to oligomers and both homo- and copolymers that contain a silicon or germanium metallole, metallaindene, and metallafluorene. The last section contains a summary and commentary on future directions.
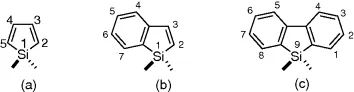
1.2 Background
The numbering scheme for siloles and the benzene-annulated derivatives is illustrated in Figure 1.1. The silaindenes are sometimes referred to as benzosiloles and the silafluorenes as dibenzosiloles, but the first name listed will be used in this chapter. The silaindene shown is technically 1H-1-silaindene and the silafluorene is 9H-9-silafluorene, but the 1H and 9H are often omitted The germanium analogs are similarly numbered and named (germaindenes and germafluorenes).
Probably the limiting factor in the assembly of the systems shown in
Figure 1.1 is the small number of C—Si bond formation methods. One of the two general methods involves a salt metathesis reaction of an organolithium or organoGrignard reagent with a silicon halide (elimination of a metal halide). The second is addition of an Si—H bond to an unsaturated organic substrate, >C=C< or —C
C—. Such a reaction is referred to as hydrosilylation and is commonly initiated with a transition metal catalyst. Nucleophilic substitutions at silicon dominate organosilicon chemistry. From an organic chemist's perspective, organosilicon chemistry is currently a matter of manipulation of single bonds. Although compounds that contain an Si=X unit (X = C, Si, N, P, transition metal) are known, their practical use in synthesis awaits future development. An exception to these general observations is the recently published transition metal-promoted reactions that can involve SiH—HC coupling for ring closure.
Simple calculations (ab initio) performed for the model silole H4C4SiH2 with a comparison with the parent carbon system, H4C4CH2, and the sulfur heterocycle, H4C4S, and also a series of N heterocycles demonstrated that the silole had the lowest HOMO–LUMO energy gap in addition to a low-lying LUMO level [2,4], The low-lying LUMO level was attributed to efficient σ*–π* overlap from the σ* orbital associated with the exocyclic σ-bonded substituents on the silicon center and the π* orbital of the butadiene unit. No such orbital is available for the parent carbon species or for the other five-membered heterocycles (with N, O or S heteroatoms). However, model systems, although simpler to calculate, do not reveal the role that substituents may play in the relative energies of the HOMO and LUMO, nor do they indicate substituent variations that might lead to better...