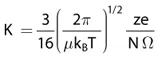Presents Practical Applications of Mass Spectrometry for Protein Analysis and Covers Their Impact on Accelerating Drug Discovery and Development
Covers both qualitative and quantitative aspects of Mass Spectrometry protein analysis in drug discovery
Principles, Instrumentation, Technologies topics include MS of peptides, proteins, and ADCs , instrumentation in protein analysis, nanospray technology in MS protein analysis, and automation in MS protein analysis
Details emerging areas from drug monitoring to patient care such as Identification and validation of biomarkers for cancer, targeted MS approaches for biomarker validation, biomarker discovery, and regulatory perspectives
Brings together the most current advances in the mass spectrometry technology and related method in protein analysis
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Chapter 1 Contemporary Protein Analysis by Ion Mobility Mass Spectrometry
Johannes P.C. Vissers and James I. Langridge
Waters Corporation, Wilmslow, UK
1.1 Introduction
The use of ion mobility as an analytical technique to detect and separate biomolecules dates back to the break of the century with the application of the method for proteomics (Valentine et al. 2006; McLean et al. 2005; Gabryelski and Froese 2003), glycomics (Taraszka et al. 2001; Jin et al. 2005; Hoaglund et al. 1997), and metabolomics (Dwivedi et al. 2008). It is a technique that separates gas-phase ions based upon their mobility in a buffer gas. This separation is related to ion size, shape, as well as m/z, and charge. The basis for separation by traditional drift tube ion mobility at a low electric limit can be derived from the Mason–Schamp equation:
where K = drift velocity vd/electric field strength E, μ = reduced mass of the ion (neutral given by (mneutralmion)/(mneutral + mion), kB = Boltzmann constant, T = temperature, z = charge state of the analyte ion, e = charge on an electron, N = number density of the drift gas, and Ω = average collision cross section. The hyphenation of ion mobility spectrometry (IMS) with MS is often referred to as ion mobility–mass spectrometry (IM–MS). The most common mass analyzer coupled with IMS comprises a time-of-flight (TOF) instrument due to the inherent high sampling rate, although other mass detection systems have been described (Kanu et al. 2008). Four different methods of ion mobility separation are currently used in combination with MS, including drift-time ion mobility spectrometry (DTIMS), aspiration ion mobility spectrometry (AIMS), differential mobility spectrometry (DMS), also called field-asymmetric waveform ion mobility spectrometry (FAIMS), and traveling-wave ion mobility spectrometry (TWIMS). A description of these methods is beyond the scope of this chapter, particularly since they have been reviewed in great detail elsewhere (Kanu et al. 2008; Lanucara et al. 2014).
The innovative demonstration of protein conformer separation by means of IMS by Clemmer et al. 1995 has prompted instrumental IM–MS designs and the broader application of IMS as an analytical tool. The designs by Pringle et al. 2007 and Baker et al. 2007, both orthogonal acceleration time-of-flight (oa-TOF) based IM–MS platform, but utilizing different IMS geometries, have been commercialized and applied for numerous applications and include drug metabolism/metabolites (Dear et al. 2010), lipids (Kliman et al. 2011), trace impurities (Eckers et al. 2007), carbohydrates (Vakhrushev et al. 2008, Schenauer et al. 2009), macromolecular protein species and viruses (Ruotolo et al. 2005, Bereszczak et al. 2014), metal-based anticancer drugs (Williams et al. 2009), and PEGylated conjugates (Bagal et al. 2008). In this chapter, the application of IMS for the identification, quantification, and characterization of proteins is illustrated by application examples that demonstrate the benefits of integrating IMS into the analytical schema in terms of increased resolution and sensitivity, as well as those obtained from collision cross section measurements.
1.2 Traveling-Wave Ion Mobility Mass Spectrometry
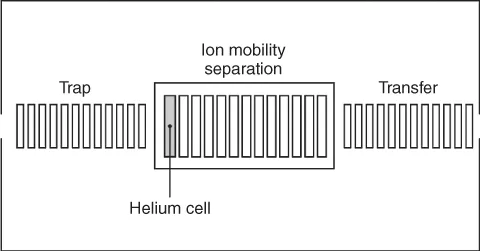
The principle of TWIMS is briefly discussed as it forms the basis of subsequent sections. A schematic of the device is shown in Figure 1.1. Details can be found in the papers of Pringle et al. 2007 and Giles et al. 2004. Ions are formed by electrospray ionization (ESI) in the source and subsequently pass through a quadrupole for mass selection before injection into the ion mobility cell. Unlike our other instruments, which use a uniform electric field across the cell for ion mobility experiments, so-called drift tube IMS, this device uses traveling-wave (T-wave) technology. The T-wave cell consists of a stacked-ring radio frequency (RF) ion guide, which incorporates a repeating sequence of transient voltages applied to the ring electrodes. These voltage pulses result in a traveling electric field that propels ions through the background gas present in the mobility cell. The total time taken for an ion to drift through the cell depends on its mobility, as well as the wave period and height, and the gas pressure. Ions with high mobility are better able to keep up with traveling waves and are pushed more quickly through the cell. Ions with low mobility crest over the waves more often and have to wait for subsequent waves to push them forward, resulting in longer drift times. To measure an arrival time distribution (ATD), ions are gated into the mobility cell using an up-front stacked-ring RF device that traps ions before releasing them into the IMS cell. The oa-TOF pulses in an asynchronous manner, sending ions that have exited the mobility cell into the TOF mass analyzer and finally to the detector. The sum of all detected ions is the ion mobility chromatogram, or mobilogram. Selecting a peak in the ion mobility chromatogram displays the underlying TOF mass spectrum. Resolution enhancements to the device are recently described (Giles et al. 2011).
Figure 1.1 Triwave ion mobility optics detail comprising a trap, helium cell, ion mobility separator and transfer.
(Source: Williams et al. 2012. Reproduced with permission of GIT.)
1.3 IM–MS and LC–IM–MS Analysis of Simple and Complex Mixtures
1.3.1 Cross Section and Structure
By measuring the mobility of an ion, information about the rotationally averaged collision cross section, which is given by shape and size, can be determined. The relationship between the mobility of an ion and its collision cross section has been derived in detail using kinetic theory (Mason and McDaniel 1988). When all experimental IM parameter values are held constant, a dependence of the ion mobility constant results only from the average cross section with K ~ 1/Ω (1996; Bowers et al./>; Henderson et al. 1999; Verbeck et al. 2002), where K = drift velocity vd/electric field strength E and Ω = average collision cross section. A detailed description of kinetic theory is beyond the scope of this discussion. Ruotolo et al. 2005 were among the first describing the use of IM–MS to decipherer protein complex structure. The analysis of the Trp RNA-binding attenuation protein (TRAP) provided compelling evidence that many features of protein assemblies, including quaternary structure, can be preserved in the absence of solvent molecules. The researchers made use of TWIMS coupled to a modified TOF mass spectrometer to measure the CCS of four charge states of an 11-mer complex, demonstrating that the lowest charge state exhibited the largest CCS, with a value in close agreement to that estimated for a ring structure determined by X-ray crystallography. To investigate the sensitivity of the various conformers to changes in internal energy, they examined collision cross sections of the apo TRAP complex as a function of activation energy by manipulating their acceleration in the atmospheric pressure interface of the instrument, shown in Figure 1.2. The experiment illustrated th...
Table of contents
Cover
Title Page
Copyright
Table of Contents
List of Contributors
Foreword
Preface
Chapter 1: Contemporary Protein Analysis by Ion Mobility Mass Spectrometry
Chapter 2: High-Resolution Accurate Mass Orbitrap and Its Application in Protein Therapeutics Bioanalysis
Chapter 3: Current Methods for the Characterization of Posttranslational Modifications in Therapeutic Proteins Using Orbitrap Mass Spectrometry
Chapter 4: Macro- to Micromolecular Quantitation of Proteins and Peptides by Mass Spectrometry
Chapter 5: Peptide and Protein Bioanalysis Using Integrated Column-to-Source Technology for High-Flow Nanospray
Chapter 6: Targeting the Right Protein Isoform: Mass Spectrometry-Based Proteomic Characterization of Alternative Splice Variants
Chapter 7: The Application of Immunoaffinity-Based Mass Spectrometry to Characterize Protein Biomarkers and Biotherapeutics
Chapter 8: Semiquantification and Isotyping of Antidrug Antibodies by Immunocapture-LC/MS for Immunogenicity Assessment
Chapter 9: Mass Spectrometry-Based Assay for High-Throughput and High-Sensitivity Biomarker Verification
Chapter 10: Monitoring Quality of Critical Reagents Used in Ligand Binding Assays with Liquid Chromatography Mass Spectrometry (LC–MS)
Chapter 11: Application of Liquid Chromatography-High Resolution Mass Spectrometry in the Quantification of Intact Proteins in Biological Fluids
Chapter 12: LC–MS/MS Bioanalytical Method Development Strategy for Therapeutic Monoclonal Antibodies in Preclinical Studies
Chapter 13: Generic Peptide Strategies for LC–MS/MS Bioanalysis of Human Monoclonal Antibody Drugs and Drug Candidates
Chapter 14: Mass Spectrometry-Based Methodologies for Pharmacokinetic Characterization of Antibody Drug Conjugate Candidates During Drug Development
Chapter 15: Sample Preparation Strategies for LC–MS Bioanalysis of Proteins
Chapter 16: Characterization of Protein Therapeutics by Mass Spectrometry
Index
End User License Agreement
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
Both plans are available with monthly, semester, or annual billing cycles.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go. Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Protein Analysis using Mass Spectrometry by Mike S. Lee, Qin C. Ji, Mike S. Lee,Qin C. Ji in PDF and/or ePUB format, as well as other popular books in Ciencias físicas & Espectroscopia y análisis de espectro. We have over 1.5 million books available in our catalogue for you to explore.