A comprehensive introduction for scientists engaged in new drug development, analysis, and approvals
Each year the pharmaceutical industry worldwide recruits thousands of recent science graduates—especially chemistry, analytical chemistry, pharmacy, and pharmaceutical majors—into its ranks. However, because of their limited background in pharmaceutical analysis most of those new recruits find making the transition from academia to industry very difficult. Designed to assist both recent graduates, as well as experienced chemists or scientists with limited regulatory, compendial or pharmaceutical analysis background, make that transition, Pharmaceutical Analysis for Small Molecules is a concise, yet comprehensive introduction to the drug development process and analysis of chemically synthesized, small molecule drugs. It features contributions by distinguished experts in the field, including editor and author, Dr. Behnam Davani, an analytical chemist with decades of technical management and teaching experience in compendial, regulatory, and industry.
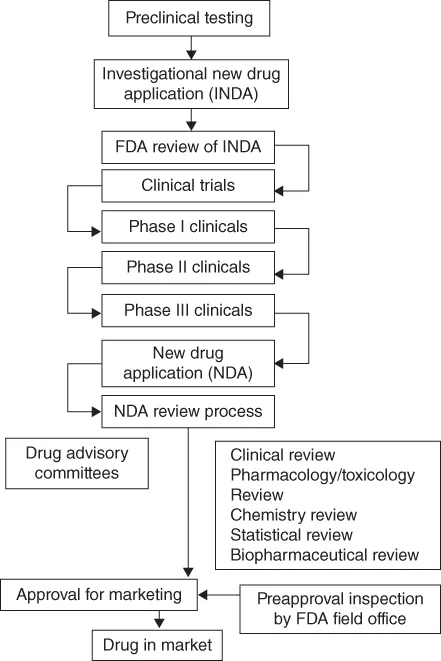
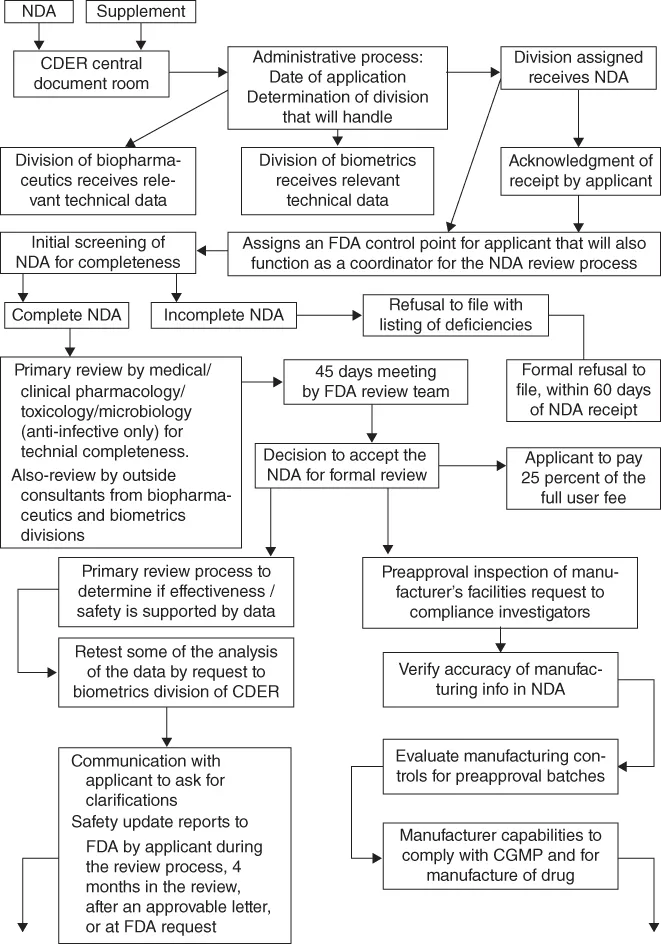
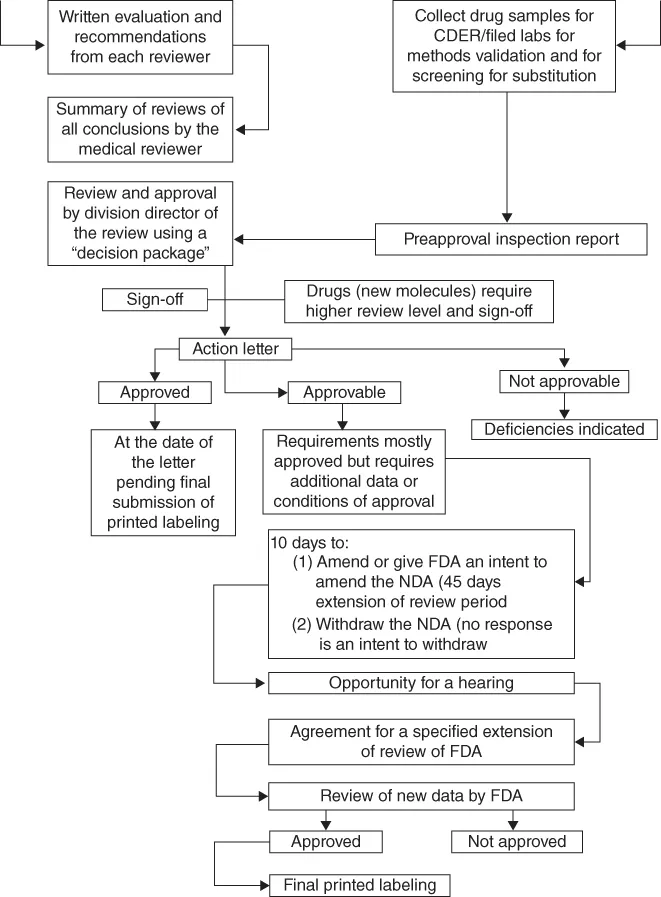
This book provides an introduction to pharmaceutical analysis for small molecules (non-biologics) using commonly used techniques for drug characterization and performance tests. The driving force for industry to perform pharmaceutical analyses is submission of such data and supporting documents to regulatory bodies for drug approval in order to market their products. In addition, related required supporting studies including good laboratory/documentation practices including analytical instrument qualification are highlighted in this book.
Topics covered include:
- Drug Approval Process and Regulatory Requirements (private standards)
- Pharmacopeias and Compendial Approval Process (public standards)
- Common methods in pharmaceutical analysis (typically compendial)
- Common Calculations for assays and impurities and other specific tests
- Analytical Method Validation, Verification, Transfer
- Specifications including how to handle out of specification (OOS) and out of trend (OOT)
- Impurities including organic, inorganic, residual solvents and elemental impurities
- Good Documentation Practices for regulatory environment
- Management of Analytical Laboratories
- Analytical Instrument Qualifications including IQ, OQ, PQ and VQ
Due to global nature of pharmaceutical industry, other topics on both regulatory (ICH) and Compendial harmonization are also highlighted.
Pharmaceutical Analysis for Small Molecules is a valuable working resource for scientists directly or indirectly involved with the drug development process, including analytical chemists, pharmaceutical scientists, pharmacists, and quality control/quality assurance professionals. It also is an excellent text/reference for graduate students in analytical chemistry, pharmacy, pharmaceutical and regulatory sciences.