This book presents advances in the field of neuronal mitochondria – functions, relation to therapeutics, and pharmacology. For scientists and researchers in both industry and academia, this book provides detailed discussion, examples, and approaches, to illustrate the potential of mitochondria as therapeutic targets for neuronal diseases.
• Helps readers understand the regulation of mitochondrial cellular processes, such as substrate metabolism, energy production, and programmed versus sporadic cell death
• Offers insights on the development of strategies for targeted therapeutic approaches and potential personalized treatments
• Includes examples of mitochondrial drugs, development, and mitochondria-targeted approaches for more efficient treatment methods and further developments in the field
• Covers the model systems and approaches needed for the development of new drugs for the central nervous system to provide potential modern therapeutics for neurodegenerative disorders

eBook - ePub
The Functions, Disease-Related Dysfunctions, and Therapeutic Targeting of Neuronal Mitochondria
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
The Functions, Disease-Related Dysfunctions, and Therapeutic Targeting of Neuronal Mitochondria
About this book
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
SECTION II
CONTROL OF MITOCHONDRIAL SIGNALING NETWORKS
4
MITOCHONDRIAL Ca2+ TRANSPORT IN THE CONTROL OF NEURONAL FUNCTIONS: MOLECULAR AND CELLULAR MECHANISMS
Yuriy M. Usachev
Department of Pharmacology, University of Iowa College of Medicine, Iowa City, Iowa
4.1 INTRODUCTION
The ability of mitochondria to transport Ca2+ has been recognized for several decades (Carafoli, 2003; Carafoli and Lehninger, 1971; Deluca and Engstrom, 1961; Drago, Pizzo, and Pozzan, 2011; Vasington and Murphy, 1962). The fact that mitochondrial Ca2+ transport remains a focus of intensive investigation to this day reflects the importance of this process in the control of many determinants of cell life and death (Babcock and Hille, 1998; Friel, 2000; Newmeyer and Ferguson-Miller, 2003; Nicholls and Budd, 2000; Orrenius, Zhivotovsky, and Nicotera, 2003; Pozzan and Rizzuto, 2000; Thayer et al., 2002) (Fig. 4.1). In neurons, mitochondria efficiently buffer Ca2+ influx during excitation, limiting the amplitude of the Ca2+ signal. Rapid Ca2+ buffering by mitochondria is followed by a much slower Ca2+ release back to the cytosol (Bernardi, 1999; Nicholls, 2005; Thayer, Usachev, and Pottorf, 2002), and such Ca2+ cycling across the inner mitochondrial membrane markedly prolongs the cytosolic Ca2+ response by producing a so-called [Ca2+]i plateau (Figs. 4.1 and 4.2). This phenomenon occurs both in central (Medler and Gleason, 2002; White and Reynolds, 1996) and peripheral (Colegrove, Albrecht, and Friel, 2000a; Dedov and Roufogalis, 2000; Friel and Tsien, 1994; Shishkin et al., 2002; Thayer and Miller, 1990) neurons but is more pronounced in the latter.
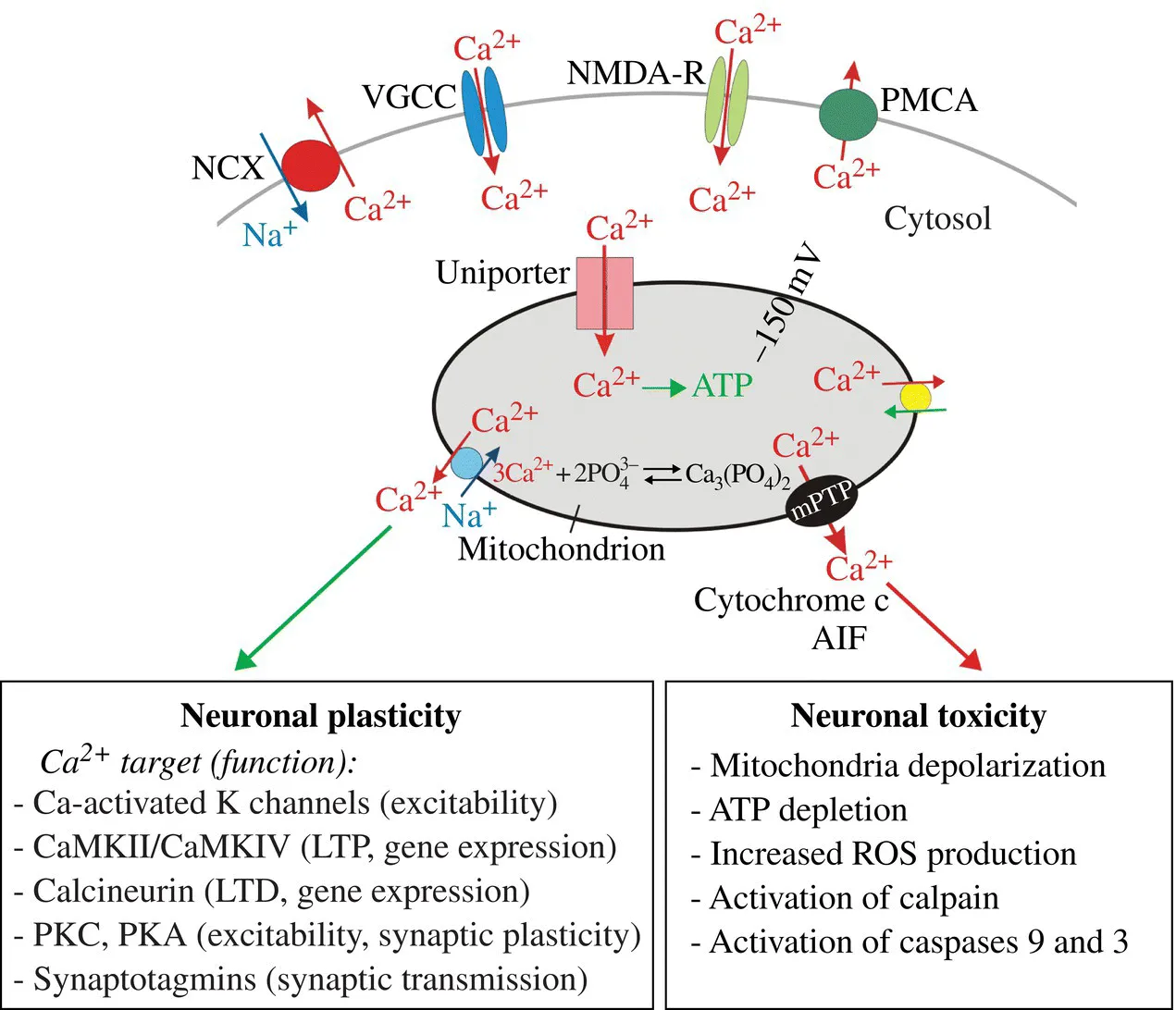
FIGURE 4.1 Mitochondrial Ca2+ signaling in neurons. Ca2+ entering neurons via voltage-gated Ca2+ channels (VGCC), glutamate NMDA receptors (NMDA-R), or other Ca2+-permeable channels (e.g., TRP channels) is rapidly taken up by mitochondria. This Ca2+ uptake is mediated by the mitochondrial Ca2+ uniporter. Within mitochondria, Ca2+ stimulates ATP synthesis through activation of several Ca2+-activated dehydrogenases (Denton, 2009; Glancy and Balaban, 2012). Free Ca2+ within the matrix is buffered into Ca-phosphate complexes. Ca2+ is released from mitochondria back into the cytosol via at least three mechanisms, Na+/Ca2+ exchanger, H+/Ca2+ exchanger, and mitochondrial permeability transition pore (mPTP). Under physiological conditions, Ca2+ mobilization from mitochondria is mediated primarily by the mitochondrial Na+/Ca2+ exchanger. The Ca2+ released from mitochondria is ultimately extruded from neurons via the plasma membrane Ca2+-ATPases (PMCA) and Na+/Ca2+ exchangers (NCX). Ca2+ uptake and release by mitochondria reduces the amplitude of increase in cytosolic Ca2+ concentration ([Ca2+]i), and prolongs the duration of [Ca2+]i elevation. By shaping [Ca2+]i, mitochondrial Ca2+ cycling modulates activity of many neuronal Ca2+ targets and influences numerous Ca2+-dependent functions such as excitability, synaptic transmission, and gene regulation. Under pathological conditions, mitochondrial overload with Ca2+ triggers opening of the mPTP, a large channel permeable for Ca2+ and small proteins including the pro-apoptotic factors cytochrome c and apoptosis-inducing factor (AIF). Opening of the mPTP triggers a cascade of toxic events (e.g., mitochondrial depolarization, collapse of ATP synthesis, increase in reactive oxygen species (ROS) production, Ca2+ deregulation, activation of caspases, and the Ca2+-dependent protease calpain), ultimately leading to neuronal demise. The deregulation of mitochondrial Ca2+ handling is implicated in neuronal toxicity in stroke, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis (ALS), and other neurodegenerative disorders (Bezprozvanny, 2009; Mattson, Gleichmann, and Cheng, 2008; Pivovarova and Andrews, 2010).
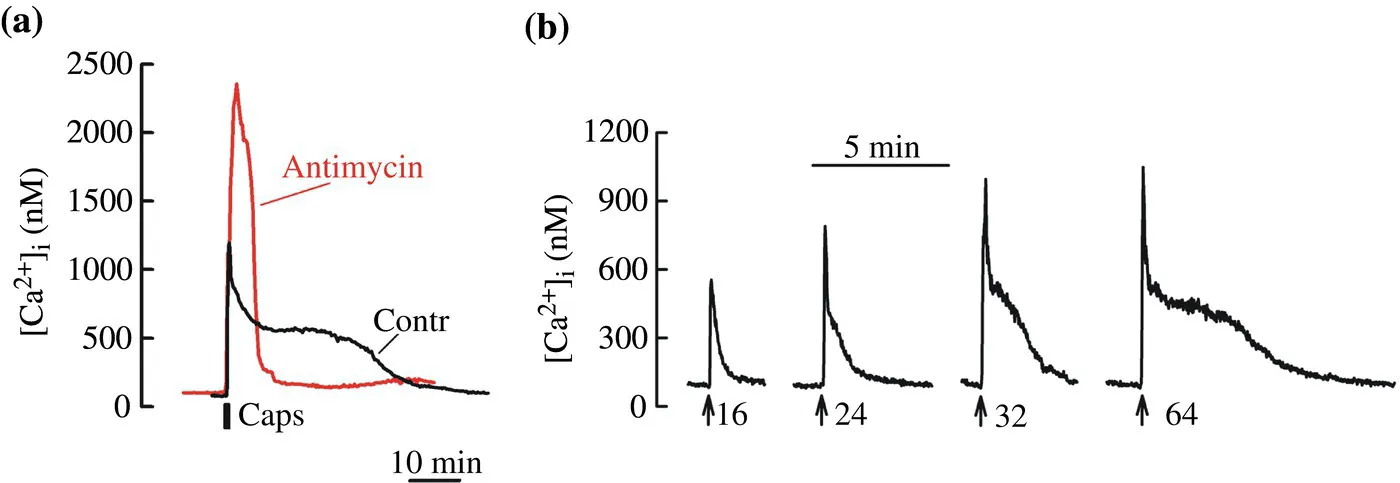
FIGURE 4.2 Mitochondrial Ca2+ cycling regulates amplitude and duration of [Ca2+]i responses in neurons. Changes in cytosolic Ca2+ concentration ([Ca2+]i) were studied in cultured dorsal root ganglion (DRG) sensory neurons using the Ca2+-sensitive indicator Fura-2 as previously described (Kim and Usachev, 2009; Kim et al., 2014; Schnizler et al., 2008; Usachev et al., 2002). a, Superimposition of [Ca2+]i responses to the TRPV1 agonist capsaicin (1 μM; 30 s) recorded in control (black) or in the presence of the electron transport inhibitor antimycin (1 μM) and ATP-synthase inhibitor oligomycin (2 μM) (grey trace). Both traces were obtained from the same cell. Inhibition of mitochondrial Ca2+ transport by the antimycin with oligomycin treatment increased the amplitude and reduced the duration of capsaicin-evoked [Ca2+]i elevation (compare grey and black traces). b, [Ca2+]i transients were evoked by trains of action potentials (8 Hz; arrows) of increased duration using extracellular field stimulation. The number of action potentials is shown under the traces. Increasing the strength of the stimulation led to a gradual appearance and prolongation of the [Ca2+]i plateau phase mediated by Ca2+ release from mitochondria.
Ca2+ transported into the mitochondrial matrix stimulates Ca2+-dependent dehydrogenases and boosts ATP production to meet increased energy demand during excitation (Denton, 2009; Glancy and Balaban, 2012; Hajnoczky et al., 1995). However, excessive load of mitochondria with Ca2+ triggers neurotoxic processes in stroke and in Alzheimer’s, Parkinson’s, and Huntington’s diseases (Bezprozvanny, 2009; Moskowitz, Lo, and Iadecola, 2010; Murphy, Fiskum, and Beal, 1999; Nicholls and Budd, 2000; Panov et al., 2002; Reynolds, 1999). In addition, mitochondrial-dependent alteration of the amplitude and duration of cytosolic Ca2+ signals influences the activation and function of numerous cytosolic Ca2+ targets, including Ca2+-activated ion channels (e.g., small- and large-conductance Ca2+-activated K+ channels, SK and BK), Ca2+ sensors at the synapse (e.g., synaptotagmins and Doc2) and numerous Ca2+-dependent enzymes (e.g., calmodulin-dependent protein kinases, protein kinases A and C, protein phosphatase 2B/calcineurin, and the Ca2+-dependent protease calpain) (Berridge, Bootman, and Lipp, 1998; Clapham, 2007; Groffen et al., 2010; Kim and Usachev, 2009; Sah, 1996; Sugita et al., 2002; Xu, Mashimo, and Sudhof, 2007; Yao et al., 2011). Accordingly, mitochondrial Ca2+ transport regulates many Ca2+-dependent functions including neuronal excitability, synaptic transmission, gene expression, and neuronal survival (see Fig. 4.1).
The fact that mitochondrial Ca2+ signaling is key to controlling neuronal life and death makes the components of mitochondrial Ca2+ transport attractive candidates for therapeutic targeting in the treatment of neurodegenerative and psychiatric disorders. Although progress in this area was long hindered by a lack of information on the identities of the molecules that mediate Ca2+ transport in and out of mitochondria, the recent discovery of several genes encoding key mitochondrial Ca2+ transporters (Marchi and Pinton, 2014; Pendin, Greotti, and Pozzan, 2014) is expected to accelerate our progress toward understanding and therapeutically exploiting this machinery.
In this chapter I will review recent advancements in the molecular biology of mitochondrial Ca2+ signaling in neurons and discuss how Ca2+ transport into and out of neuronal mitochondria contributes to the control of neuron excitability, synaptic transmission, and gene expression (see Fig. 4.1). The role of mitochondrial Ca2+ overload in neuronal toxicity, which is another important aspect of mitochondrial Ca2+ transport in neurons, is reviewed in the contribution by Brustovetsky (see Chapter 1).
4.2 PHYSIOLOGICAL AND PHARMACOLOGICAL CHARACTERISTICS OF MITOCHONDRIAL Ca2+ TRANSPORT IN NEURONS
In neurons at rest, total Ca2+ concentration in the matrix ([Ca]mt) is low, usually <100 nM (Pivovarova et al., 1999; Stanika et al., 2012). During electrical or synaptic stimulation, [Ca]mt can increase to millimolar levels within seconds (Pivovarova et al., 1999, 2002). In the case of modest to strong stimulation, the rate of mitochondrial Ca2+ uptake in intact neur...
Table of contents
- COVER
- TITLE PAGE
- TABLE OF CONTENTS
- CONTRIBUTORS
- PREFACE
- SECTION I: MITOCHONDRIAL STRUCTURE AND ION CHANNELS
- SECTION II: CONTROL OF MITOCHONDRIAL SIGNALING NETWORKS
- SECTION III: DEFECTIVE MITOCHONDRIAL DYNAMICS AND MITOPHAGY
- SECTION IV: MITOCHONDRIA-TARGETED THERAPEUTICS AND MODEL SYSTEMS
- INDEX
- END USER LICENSE AGREEMENT
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access The Functions, Disease-Related Dysfunctions, and Therapeutic Targeting of Neuronal Mitochondria by J. Marie Hardwick, Valentin K. Gribkoff,Elizabeth A. Jonas, Valentin K. Gribkoff, Elizabeth A. Jonas in PDF and/or ePUB format, as well as other popular books in Scienze biologiche & Farmacologia. We have over 1.5 million books available in our catalogue for you to explore.